T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis

 and
and 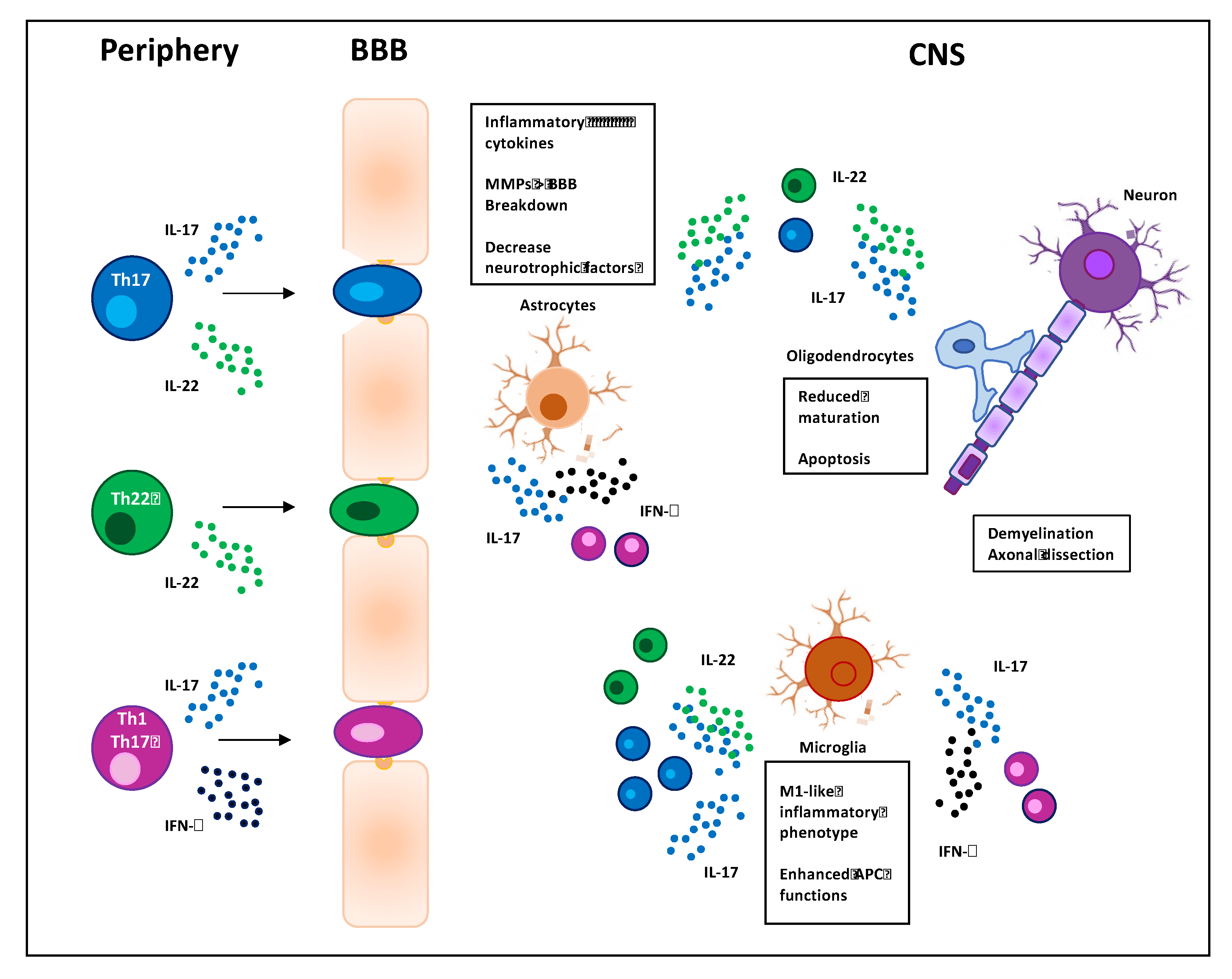{kind=link}
Abstract
:1. Introduction
2. Th Cell Subsets
2.1. Th1 Cells
2.2. Th17 Cells
2.3. Th1-Like Th17 Cells
2.4. Th22 Cells
2.5. Th9 Cells
3. Treg Cells
4. T Cell Targeting Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Brownlee, W.J.; Hardy, T.A.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2017, 389, 1336–1346. [Google Scholar] [CrossRef]
- Lorscheider, J.; Buzzard, K.; Jokubaitis, V.; Spelman, T.; Havrdova, E.; Horakova, D.; Trojano, M.; Izquierdo, G.; Girard, M.; Duquette, P.; et al. Defining secondary progressive multiple sclerosis. Brain 2016, 139, 2395–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple sclerosis: Mechanisms and immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Hafler, D.A.; Benjamin, D.S.; Burks, J.; Weiner, H.L. Myelin basic protein and proteolipid protein reactivity of brain- and cerebrospinal fluid-derived T cell clones in multiple sclerosis and postinfectious encephalomyelitis. J. Immunol. 1987, 139, 68–72. [Google Scholar]
- Ota, K.; Matsui, M.; Milford, E.L.; Mackin, G.A.; Weiner, H.L.; Hafler, D.A. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature 1990, 346, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Zamvil, S.; Nelson, P.; Trotter, J.; Mitchell, D.; Knobler, R.; Fritz, R.; Steinman, L. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature 1985, 317, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Zamvil, S.S.; Nelson, P.A.; Mitchell, D.J.; Knobler, R.L.; Fritz, R.B.; Steinman, L. Encephalitogenic T cell clones specific for myelin basic protein. An unusual bias in antigen recognition. J. Exp. Med. 1985, 162, 2107–2124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Markovic-Plese, S.; Lacet, B.; Raus, J.; Weiner, H.L.; Hafler, D.A. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J. Exp. Med. 1994, 179, 973–984. [Google Scholar] [CrossRef]
- Batoulis, H.; Addicks, K.; Kuerten, S. Emerging concepts in autoimmune encephalomyelitis beyond the CD4/T(h)1 paradigm. Ann. Anat. 2010, 192, 179–193. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics, C.; Wellcome Trust Case Control, C.; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- Sallusto, F. Heterogeneity of human CD4(+) T cells against microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytometry A 2014, 85, 36–42. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Mosmann, T.R.; Coffman, R.L. Th1 and Th2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Human Th1 dichotomy: Origin, phenotype and biologic activities. Immunology 2014, 144, 343–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.S.; Macatonia, S.E.; Tripp, C.S.; Wolf, S.F.; O’Garra, A.; Murphy, K.M. Development of Th1 CD4+ T cells through IL-12 produced by listeria-induced macrophages. Science 1993, 260, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Ando, D.G.; Clayton, J.; Kono, D.; Urban, J.L.; Sercarz, E.E. Encephalitogenic T cells in the B10.Pl model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989, 124, 132–143. [Google Scholar] [CrossRef]
- Voskuhl, R.R.; Martin, R.; Bergman, C.; Dalal, M.; Ruddle, N.H.; McFarland, H.F. T helper 1 (Th1) functional phenotype of human myelin basic protein-specific T lymphocytes. Autoimmunity 1993, 15, 137–143. [Google Scholar] [CrossRef]
- Merrill, J.E.; Kono, D.H.; Clayton, J.; Ando, D.G.; Hinton, D.R.; Hofman, F.M. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.Pl mice. Proc. Natl. Acad. Sci. USA 1992, 89, 574–578. [Google Scholar] [CrossRef] [Green Version]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Baron, J.L.; Madri, J.A.; Ruddle, N.H.; Hashim, G.; Janeway, C.A., Jr. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J. Exp. Med. 1993, 177, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Segal, B.M.; Shevach, E.M. IL-12 unmasks latent autoimmune disease in resistant mice. J. Exp. Med. 1996, 184, 771–775. [Google Scholar] [CrossRef] [Green Version]
- Panitch, H.S.; Hirsch, R.L.; Haley, A.S.; Johnson, K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hirsch, R.L.; Schindler, J.; Johnson, K.P. Treatment of multiple sclerosis with gamma interferon: Exacerbations associated with activation of the immune system. Neurology 1987, 37, 1097–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the cns during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Lohr, K.; Floess, S.; Zimmermann, J.; Ulrich, R.; Gudi, V.; Beineke, A.; Baumgartner, W.; Muller, M.; Huehn, J.; et al. Effector molecules released by Th1 but not Th17 cells drive an M1 response in microglia. Brain Behav. Immun. 2014, 37, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Gran, B.; Chu, N.; Zhang, G.X.; Yu, S.; Li, Y.; Chen, X.H.; Kamoun, M.; Rostami, A. Early administration of IL-12 suppresses eae through induction of interferon-gamma. J. Neuroimmunol. 2004, 156, 123–131. [Google Scholar] [CrossRef]
- Becher, B.; Durell, B.G.; Noelle, R.J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Invest. 2002, 110, 493–497. [Google Scholar] [CrossRef]
- Zhang, G.X.; Gran, B.; Yu, S.; Li, J.; Siglienti, I.; Chen, X.; Kamoun, M.; Rostami, A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J. Immunol. 2003, 170, 2153–2160. [Google Scholar] [CrossRef]
- Ferber, I.A.; Brocke, S.; Taylor-Edwards, C.; Ridgway, W.; Dinisco, C.; Steinman, L.; Dalton, D.; Fathman, C.G. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (eae). J. Immunol. 1996, 156, 5–7. [Google Scholar]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [Green Version]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Veldhoen, M. Differentiation and function of Th17 T cells. Curr. Opin. Immunol. 2007, 19, 281–286. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Defining the human T helper 17 cell phenotype. Trends Immunol. 2012, 33, 505–512. [Google Scholar] [CrossRef]
- Kolls, J.K.; Linden, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef]
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Battistini, L.; Borsellino, G. Advances in T helper 17 cell biology: Pathogenic role and potential therapy in multiple sclerosis. Mediators Inflamm. 2015, 2015, 475158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahmasebinia, F.; Pourgholaminejad, A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Kronisch, J.; Khorooshi, R.; Knier, B.; Toft-Hansen, H.; Gudi, V.; Floess, S.; Huehn, J.; Owens, T.; Korn, T.; et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflammation 2017, 14, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, J.; Ciric, B.; Marek, R.; Sadhukhan, S.; Caruso, M.L.; Shafagh, J.; Fitzgerald, D.C.; Shindler, K.S.; Rostami, A. Functional interleukin-17 receptor a is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J. Neuroinflammation 2009, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elain, G.; Jeanneau, K.; Rutkowska, A.; Mir, A.K.; Dev, K.K. The selective anti-IL17A monoclonal antibody secukinumab (AIN457) attenuates IL17A-induced levels of IL6 in human astrocytes. Glia 2014, 62, 725–735. [Google Scholar] [CrossRef]
- Kang, Z.; Altuntas, C.Z.; Gulen, M.F.; Liu, C.; Giltiay, N.; Qin, H.; Liu, L.; Qian, W.; Ransohoff, R.M.; Bergmann, C.; et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 2010, 32, 414–425. [Google Scholar] [CrossRef] [Green Version]
- Prajeeth, C.K.; Dittrich-Breiholz, O.; Talbot, S.R.; Robert, P.A.; Huehn, J.; Stangel, M. Ifn-gamma producing Th1 cells induce different transcriptional profiles in microglia and astrocytes. Front. Cell Neurosci. 2018, 12, 352. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Paintlia, M.K.; Paintlia, A.S.; Singh, A.K.; Singh, I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J. Neurochem. 2011, 116, 508–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulamea, A.O. Role of oligodendrocyte dysfunction in demyelination, remyelination and neurodegeneration in multiple sclerosis. Adv. Exp. Med. Biol. 2017, 958, 91–127. [Google Scholar] [PubMed]
- Stromnes, I.M.; Cerretti, L.M.; Liggitt, D.; Harris, R.A.; Goverman, J.M. Differential regulation of central nervous system autoimmunity by T(h)1 and T(h)17 cells. Nat. Med. 2008, 14, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Passos, G.R.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 cells pathways in multiple sclerosis and neuromyelitis optica spectrum disorders: Pathophysiological and therapeutic implications. Mediators Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef] [PubMed]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Matusevicius, D.; Kivisakk, P.; He, B.; Kostulas, N.; Ozenci, V.; Fredrikson, S.; Link, H. Interleukin-17 mrna expression in blood and csf mononuclear cells is augmented in multiple sclerosis. Mult. Scler. 1999, 5, 101–104. [Google Scholar] [CrossRef]
- Durelli, L.; Conti, L.; Clerico, M.; Boselli, D.; Contessa, G.; Ripellino, P.; Ferrero, B.; Eid, P.; Novelli, F. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann. Neurol. 2009, 65, 499–509. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, C.J.; Krakauer, M.; Bendtzen, K.; Lund, H.; Sellebjerg, F.; Nielsen, C.H. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology 2008, 125, 161–169. [Google Scholar] [CrossRef]
- Arellano, G.; Acuna, E.; Reyes, L.I.; Ottum, P.A.; De Sarno, P.; Villarroel, L.; Ciampi, E.; Uribe-San Martin, R.; Carcamo, C.; Naves, R. Th1 and Th17 cells and associated cytokines discriminate among clinically isolated syndrome and multiple sclerosis phenotypes. Front. Immunol. 2017, 8, 753. [Google Scholar] [CrossRef]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human Th17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiadi, A.F.; Abbas, A.R.; Jeet, S.; Wong, K.; Bischof, A.; Peng, I.; Lee, J.; Bremer, M.; Eggers, E.L.; DeVoss, J.; et al. IL-17A is associated with the breakdown of the blood-brain barrier in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2019, 332, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanke, F.; Tang, Y.; Gronke, K.; Klebow, S.; Moos, S.; Hauptmann, J.; Shanmugavadivu, A.; Regen, T.; Mufazalov, I.A.; Gabriel, L.A.; et al. Expression of IL-17F is associated with non-pathogenic Th17 cells. J. Mol. Med. (Berl) 2018, 96, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Ghosh, C.; Hossain, M.; Linfield, D.; Rezaee, F.; Janigro, D.; Marchi, N.; van Boxel-Dezaire, A.H.H. IFN-gamma, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: Relevance for neuro-inflammatory diseases. Biochem. Biophys. Res. Commun. 2018, 507, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Colamatteo, A.; Maggioli, E.; Azevedo Loiola, R.; Hamid Sheikh, M.; Cali, G.; Bruzzese, D.; Maniscalco, G.T.; Centonze, D.; Buttari, F.; Lanzillo, R.; et al. Reduced annexin A1 expression associates with disease severity and inflammation in multiple sclerosis patients. J. Immunol. 2019, 203, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory il-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in Th17 development and Th17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [Green Version]
- Whitley, S.K.; Balasubramani, A.; Zindl, C.; Sen, R.; Shibata, Y.; Crawford, G.E.; Weathington, N.M.; Hatton, R.D.; Weaver, C.T. IL-1R signaling promotes STAT33 and NF-kappaB factor recruitment to distal cis-regulatory elements that regulate IL17A/F transcription. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Ghoreschi, K.; Laurence, A.; Yang, X.P.; Kanno, Y.; O’Shea, J.J. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010, 21, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Cosmi, L.; De Palma, R.; Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Rodolico, G.; Querci, V.; Abbate, G.; Angeli, R.; et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J. Exp. Med. 2008, 205, 1903–1916. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Human Th17 cells: Are they different from murine Th17 cells? Eur. J. Immunol. 2009, 39, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector Th17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef]
- Basu, R.; Whitley, S.K.; Bhaumik, S.; Zindl, C.L.; Schoeb, T.R.; Benveniste, E.N.; Pear, W.S.; Hatton, R.D.; Weaver, C.T. IL-1 signaling modulates activation of stat transcription factors to antagonize retinoic acid signaling and control the Th17 cell-iTreg cell balance. Nat. Immunol. 2015, 16, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Mailer, R.K.; Joly, A.L.; Liu, S.; Elias, S.; Tegner, J.; Andersson, J. IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci. Rep. 2015, 5, 14674. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human T(h)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupe, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(h)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-beta are required for differentiation of human T(h)17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef]
- Porciello, N.; Grazioli, P.; Campese, A.F.; Kunkl, M.; Caristi, S.; Mastrogiovanni, M.; Muscolini, M.; Spadaro, F.; Favre, C.; Nunes, J.A.; et al. A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscolini, M.; Camperio, C.; Porciello, N.; Caristi, S.; Capuano, C.; Viola, A.; Galandrini, R.; Tuosto, L. Phosphatidylinositol 4-phosphate 5-kinase alpha and Vav1 mutual cooperation in CD28-mediated actin remodeling and signaling functions. J. Immunol. 2015, 194, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porciello, N.; Tuosto, L. Cd28 costimulatory signals in T lymphocyte activation: Emerging functions beyond a qualitative and quantitative support to TCR signalling. Cytokine Growth Factor Rev. 2016, 28, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Porciello, N.; Kunkl, M.; Tuosto, L. CD28 between tolerance and autoimmunity: The side effects of animal models. F1000Research 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Santarlasci, V.; Maggi, L.; Capone, M.; Querci, V.; Beltrame, L.; Cavalieri, D.; D’Aiuto, E.; Cimaz, R.; Nebbioso, A.; Liotta, F.; et al. Rarity of human T helper 17 cells is due to retinoic acid orphan receptor-dependent mechanisms that limit their expansion. Immunity 2012, 36, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Kunkl, M.; Porciello, N.; Mastrogiovanni, M.; Capuano, C.; Lucantoni, F.; Moretti, C.; Persson, J.L.; Galandrini, R.; Buzzetti, R.; Tuosto, L. ISA-2011B, a phosphatidylinositol 4-phosphate 5-kinase alpha inhibitor, impairs CD28-dependent costimulatory and pro-inflammatory signals in human T lymphocytes. Front. Immunol. 2017, 8, 502. [Google Scholar] [CrossRef] [Green Version]
- Kunkl, M.; Sambucci, M.; Ruggieri, S.; Amormino, C.; Tortorella, C.; Gasperini, C.; Battistini, L.; Tuosto, L. CD28 autonomous signaling up-regulates c-myc expression and promotes glycolysis enabling inflammatory T cell responses in multiple sclerosis. Cells 2019, 8, 575. [Google Scholar] [CrossRef] [Green Version]
- Camperio, C.; Muscolini, M.; Volpe, E.; Di Mitri, D.; Mechelli, R.; Buscarinu, M.C.; Ruggieri, S.; Piccolella, E.; Salvetti, M.; Gasperini, C.; et al. CD28 ligation in the absence of TCR stimulation up-regulates IL-17A and pro-inflammatory cytokines in relapsing-remitting multiple sclerosis T lymphocytes. Immunol. Lett. 2014, 158, 134–142. [Google Scholar] [CrossRef]
- Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Annunziato, F. Th17 plasticity: Pathophysiology and treatment of chronic inflammatory disorders. Curr. Opin. Pharmacol. 2014, 17, 12–16. [Google Scholar] [CrossRef]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Capone, A.; Bianco, M.; Ruocco, G.; De Bardi, M.; Battistini, L.; Ruggieri, S.; Gasperini, C.; Centonze, D.; Sette, C.; Volpe, E. Distinct expression of inflammatory features in T helper 17 cells from multiple sclerosis patients. Cells 2019, 8, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, D.; Notarbartolo, S.; Croonenborghs, T.; Patel, B.; Cialic, R.; Yang, T.H.; Aschenbrenner, D.; Andersson, K.M.; Gattorno, M.; Pham, M.; et al. Transcriptional signature of human pro-inflammatory Th17 cells identifies reduced IL10 gene expression in multiple sclerosis. Nat. Commun. 2017, 8, 1600. [Google Scholar] [CrossRef]
- Ghezzi, L.; Cantoni, C.; Cignarella, F.; Bollman, B.; Cross, A.H.; Salter, A.; Galimberti, D.; Cella, M.; Piccio, L. T cells producing GM-CSF and IL-13 are enriched in the cerebrospinal fluid of relapsing MS patients. Mult. Scler. 2019, 1352458519852092. [Google Scholar] [CrossRef]
- Muls, N.; Nasr, Z.; Dang, H.A.; Sindic, C.; van Pesch, V. IL-22, GM-CSF and IL-17 In peripheral CD4+ T cell subpopulations during multiple sclerosis relapses and remission. Impact of corticosteroid therapy. PLoS ONE 2017, 12, e0173780. [Google Scholar] [CrossRef] [Green Version]
- Trifari, S.; Spits, H. IL-22-producing CD4+ T cells: Middle-men between the immune system and its environment. Eur J. Immunol. 2010, 40, 2369–2371. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Cavani, A.; Schmidt-Weber, C. IL-17 and IL-22: Siblings, not twins. Trends Immunol. 2010, 31, 354–361. [Google Scholar] [CrossRef]
- Shabgah, A.G.; Navashenaq, J.G.; Shabgah, O.G.; Mohammadi, H.; Sahebkar, A. Interleukin-22 in human inflammatory diseases and viral infections. Autoimmun. Rev. 2017, 16, 1209–1218. [Google Scholar] [CrossRef]
- Xin, N.; Namaka, M.P.; Dou, C.; Zhang, Y. Exploring the role of interleukin-22 in neurological and autoimmune disorders. Int. Immunopharmacol. 2015, 28, 1076–1083. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(h)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Duhen, T.; Campbell, D.J. Il-1beta promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014, 193, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Cimaz, R.; Capone, M.; Santarlasci, V.; Querci, V.; Simonini, G.; Nencini, F.; Liotta, F.; Romagnani, S.; Maggi, E.; et al. Brief report: Etanercept inhibits the tumor necrosis factor alpha-driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol 2014, 66, 1372–1377. [Google Scholar] [CrossRef]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic Th17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Segal, B.M. T(h)17 cytokines in autoimmune neuro-inflammation. Curr. Opin. Immunol. 2011, 23, 707–712. [Google Scholar] [CrossRef] [Green Version]
- Kebir, H.; Ifergan, I.; Alvarez, J.I.; Bernard, M.; Poirier, J.; Arbour, N.; Duquette, P.; Prat, A. Preferential recruitment of interferon-gamma-expressing Th17 cells in multiple sclerosis. Ann. Neurol. 2009, 66, 390–402. [Google Scholar] [CrossRef]
- Edwards, L.J.; Robins, R.A.; Constantinescu, C.S. Th17/Th1 phenotype in demyelinating disease. Cytokine 2010, 50, 19–23. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra274. [Google Scholar] [CrossRef] [Green Version]
- Paroni, M.; Maltese, V.; De Simone, M.; Ranzani, V.; Larghi, P.; Fenoglio, C.; Pietroboni, A.M.; De Riz, M.A.; Crosti, M.C.; Maglie, S.; et al. Recognition of viral and self-antigens by Th1 and Th1/Th17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J. Allergy Clin. Immunol. 2017, 140, 797–808. [Google Scholar] [CrossRef] [Green Version]
- van Langelaar, J.; van der Vuurst de Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.; de Vries, H.E.; Lubberts, E.; et al. T helper 17.1 cells associate with multiple sclerosis disease activity: Perspectives for early intervention. Brain 2018, 141, 1334–1349. [Google Scholar] [CrossRef]
- Dumoutier, L.; Van Roost, E.; Ameye, G.; Michaux, L.; Renauld, J.C. IL-TIF/IL-22: Genomic organization and mapping of the human and mouse genes. Genes Immun. 2000, 1, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef]
- Trifari, S.; Kaplan, C.D.; Tran, E.H.; Crellin, N.K.; Spits, H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(h)-17, T(h)1 and T(h)2 cells. Nat. Immunol. 2009, 10, 864–871. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Invest. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Witte, E.; Witte, K.; Warszawska, K.; Sabat, R.; Wolk, K. Interleukin-22: A cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010, 21, 365–379. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, F.; Arasteh, J.M.; Lawley, T.D. Pathogen resistance mediated by IL-22 signaling at the epithelial-microbiota interface. J. Mol. Biol. 2015, 427, 3676–3682. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Li, R.; Dai, Y.; Wu, A.; Wang, H.; Cheng, C.; Qiu, W.; Lu, Z.; Zhong, X.; Shu, Y.; et al. IL-22 secreting CD4+ T cells in the patients with neuromyelitis optica and multiple sclerosis. J. Neuroimmunol. 2013, 261, 87–91. [Google Scholar] [CrossRef]
- Perriard, G.; Mathias, A.; Enz, L.; Canales, M.; Schluep, M.; Gentner, M.; Schaeren-Wiemers, N.; Du Pasquier, R.A. Interleukin-22 is increased in multiple sclerosis patients and targets astrocytes. J. Neuroinflammation 2015, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Wing, A.C.; Hygino, J.; Ferreira, T.B.; Kasahara, T.M.; Barros, P.O.; Sacramento, P.M.; Andrade, R.M.; Camargo, S.; Rueda, F.; Alves-Leon, S.V.; et al. Interleukin-17- and interleukin-22-secreting myelin-specific CD4(+) T cells resistant to corticoids are related with active brain lesions in multiple sclerosis patients. Immunology 2016, 147, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Almolda, B.; Costa, M.; Montoya, M.; Gonzalez, B.; Castellano, B. Increase in Th17 and T-reg lymphocytes and decrease of IL22 correlate with the recovery phase of acute EAE in rat. PLoS ONE 2011, 6, e27473. [Google Scholar] [CrossRef] [Green Version]
- Rolla, S.; Bardina, V.; De Mercanti, S.; Quaglino, P.; De Palma, R.; Gned, D.; Brusa, D.; Durelli, L.; Novelli, F.; Clerico, M. Th22 cells are expanded in multiple sclerosis and are resistant to IFN-beta. J. Leukoc. Biol. 2014, 96, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Zhen, J.; Yuan, J.; Fu, Y.; Zhu, R.; Wang, M.; Chang, H.; Zhao, Y.; Wang, D.; Lu, Z. Il-22 promotes fas expression in oligodendrocytes and inhibits FOXP3 expression in T cells by activating the NF-kappaB pathway in multiple sclerosis. Mol. Immunol. 2017, 82, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Beyeen, A.D.; Adzemovic, M.Z.; Ockinger, J.; Stridh, P.; Becanovic, K.; Laaksonen, H.; Lassmann, H.; Harris, R.A.; Hillert, J.; Alfredsson, L.; et al. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J. Immunol. 2010, 185, 6883–6890. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, H.; Guerreiro-Cacais, A.O.; Adzemovic, M.Z.; Parsa, R.; Zeitelhofer, M.; Jagodic, M.; Olsson, T. The multiple sclerosis risk gene IL22RA2 contributes to a more severe murine autoimmune neuroinflammation. Genes Immun. 2014, 15, 457–465. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced FOXP3+ T cells and, together with TGF-beta, generates iL-9+ IL-10+ FOXP3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Jager, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 2009, 183, 7169–7177. [Google Scholar] [CrossRef]
- Elyaman, W.; Bradshaw, E.M.; Uyttenhove, C.; Dardalhon, V.; Awasthi, A.; Imitola, J.; Bettelli, E.; Oukka, M.; van Snick, J.; Renauld, J.C.; et al. IL-9 induces differentiation of Th17 cells and enhances function of FOXP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12885–12890. [Google Scholar] [CrossRef] [Green Version]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.E.; Ramos, H.L.; Wei, L.; Davidson, T.S.; Bouladoux, N.; et al. Generation of pathogenic T(h)17 cells in the absence of TGF-beta signalling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Nourbakhsh, B.; Ciric, B.; Zhang, G.X.; Rostami, A. Neutralization of IL-9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J. Immunol. 2010, 185, 4095–4100. [Google Scholar] [CrossRef] [Green Version]
- Nowak, E.C.; Weaver, C.T.; Turner, H.; Begum-Haque, S.; Becher, B.; Schreiner, B.; Coyle, A.J.; Kasper, L.H.; Noelle, R.J. IL-9 as a mediator of Th17-driven inflammatory disease. J. Exp. Med. 2009, 206, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, G.; Rossi, S.; Motta, C.; Macchiarulo, G.; Barbieri, F.; De Bardi, M.; Borsellino, G.; Finardi, A.; Grasso, M.G.; Ruggieri, S.; et al. T helper 9 cells induced by plasmacytoid dendritic cells regulate interleukin-17 in multiple sclerosis. Clin. Sci. (Lond) 2015, 129, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Tateishi, T.; Isobe, N.; Yonekawa, T.; Yamasaki, R.; Matsuse, D.; Murai, H.; Kira, J. Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS ONE 2013, 8, e61835. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, R.H.; Cases, O.; Lelievre, V.; Mesples, B.; Renauld, J.C.; Loron, G.; Degos, V.; Dournaud, P.; Baud, O.; Gressens, P. IL-9/IL-9 receptor signaling selectively protects cortical neurons against developmental apoptosis. Cell Death Differ. 2008, 15, 1542–1552. [Google Scholar] [CrossRef] [Green Version]
- Bsibsi, M.; Persoon-Deen, C.; Verwer, R.W.; Meeuwsen, S.; Ravid, R.; Van Noort, J.M. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53, 688–695. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. FOXP3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Povoleri, G.A.; Scotta, C.; Nova-Lamperti, E.A.; John, S.; Lombardi, G.; Afzali, B. Thymic versus induced regulatory T cells - who regulates the regulators? Front. Immunol. 2013, 4, 169. [Google Scholar] [CrossRef] [Green Version]
- Sambucci, M.; Gargano, F.; Guerrera, G.; Battistini, L.; Borsellino, G. One, no one, and one hundred thousand: T regulatory cells’ multiple identities in neuroimmunity. Front. Immunol. 2019, 10, 2947. [Google Scholar] [CrossRef]
- Carbone, F.; De Rosa, V.; Carrieri, P.B.; Montella, S.; Bruzzese, D.; Porcellini, A.; Procaccini, C.; La Cava, A.; Matarese, G. Regulatory T cell proliferative potential is impaired in human autoimmune disease. Nat. Med. 2014, 20, 69. [Google Scholar] [CrossRef]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174. [Google Scholar] [CrossRef] [Green Version]
- Mirandola, S.R.; Hallal, D.E.; Farias, A.S.; Oliveira, E.C.; Brandao, C.O.; Ruocco, H.H.; Damasceno, B.P.; Santos, L.M. Interferon-beta modifies the peripheral blood cell cytokine secretion in patients with multiple sclerosis. Int. Immunopharmacol. 2009, 9, 824–830. [Google Scholar] [CrossRef]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological aspects of approved MS therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef]
- Chen, M.; Chen, G.; Deng, S.; Liu, X.; Hutton, G.J.; Hong, J. IFN-beta induces the proliferation of CD4+CD25+FOXP3+ regulatory T cells through upregulation of GITRL on dendritic cells in the treatment of multiple sclerosis. J. Neuroimmunol. 2012, 242, 39–46. [Google Scholar] [CrossRef]
- Abdel-Dayem, M.A.; Shaker, M.E.; Gameil, N.M. Impact of interferon beta-1b, interferon beta-1a and fingolimod therapies on serum interleukins-22, 32alpha and 34 concentrations in patients with relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2019, 337, 577062. [Google Scholar] [CrossRef]
- Weber, M.S.; Starck, M.; Wagenpfeil, S.; Meinl, E.; Hohlfeld, R.; Farina, C. Multiple sclerosis: Glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004, 127, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ifergan, I.; Antel, J.P.; Seguin, R.; Duddy, M.; Lapierre, Y.; Jalili, F.; Bar-Or, A. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J. Immunol. 2004, 172, 7144–7153. [Google Scholar] [CrossRef] [Green Version]
- Vieira, P.L.; Heystek, H.C.; Wormmeester, J.; Wierenga, E.A.; Kapsenberg, M.L. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J. Immunol. 2003, 170, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, O.; Farina, C.; Yassouridis, A.; Wiendl, H.; Then Bergh, F.; Dose, T.; Wekerle, H.; Hohlfeld, R. Multiple sclerosis: Comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7452–7457. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Gran, B.; Costello, K.; Johnson, K.; Martin, R.; Dhib-Jalbut, S. Glatiramer acetate induces a Th2-biased response and crossreactivity with myelin basic protein in patients with MS. Mult. Scler. 2001, 7, 209–219. [Google Scholar] [CrossRef]
- Hong, J.; Li, N.; Zhang, X.; Zheng, B.; Zhang, J.Z. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor FOXP3. Proc. Natl. Acad. Sci. USA 2005, 102, 6449–6454. [Google Scholar] [CrossRef] [Green Version]
- Spadaro, M.; Montarolo, F.; Perga, S.; Martire, S.; Brescia, F.; Malucchi, S.; Bertolotto, A. Biological activity of glatiramer acetate on Treg and anti-inflammatory monocytes persists for more than 10years in responder multiple sclerosis patients. Clin. Immunol. 2017, 181, 83–88. [Google Scholar] [CrossRef]
- Wolinsky, J.S.; Narayana, P.A.; O’Connor, P.; Coyle, P.K.; Ford, C.; Johnson, K.; Miller, A.; Pardo, L.; Kadosh, S.; Ladkani, D.; et al. Glatiramer acetate in primary progressive multiple sclerosis: Results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann. Neurol. 2007, 61, 14–24. [Google Scholar] [CrossRef]
- Correale, J.; Gaitan, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [Green Version]
- Ford, C.; Goodman, A.D.; Johnson, K.; Kachuck, N.; Lindsey, J.W.; Lisak, R.; Luzzio, C.; Myers, L.; Panitch, H.; Preiningerova, J.; et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: Results from the 15-year analysis of the us prospective open-label study of glatiramer acetate. Mult. Scler. 2010, 16, 342–350. [Google Scholar] [CrossRef]
- Dubey, D.; Kieseier, B.C.; Hartung, H.P.; Hemmer, B.; Warnke, C.; Menge, T.; Miller-Little, W.A.; Stuve, O. Dimethyl fumarate in relapsing-remitting multiple sclerosis: Rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Exp. Rev. Neurother. 2015, 15, 339–346. [Google Scholar] [CrossRef]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflammation 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- McGuire, V.A.; Ruiz-Zorrilla Diez, T.; Emmerich, C.H.; Strickson, S.; Ritorto, M.S.; Sutavani, R.V.; Weibeta, A.; Houslay, K.F.; Knebel, A.; Meakin, P.J.; et al. Dimethyl fumarate blocks pro-inflammatory cytokine production via inhibition of TLR induced M1 and K63 ubiquitin chain formation. Sci. Rep. 2016, 6, 31159. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Rezk, A.; Ghadiri, M.; Luessi, F.; Zipp, F.; Li, H.; Giacomini, P.S.; Antel, J.; Bar-Or, A. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J. Immunol. 2017, 198, 691–698. [Google Scholar] [CrossRef]
- Gillard, G.O.; Collette, B.; Anderson, J.; Chao, J.; Scannevin, R.H.; Huss, D.J.; Fontenot, J.D. Dmf, but not other fumarates, inhibits NF-kappaB activity in vitro in an NRF2-independent manner. J. Neuroimmunol. 2015, 283, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Ghadiri, M.; Rezk, A.; Li, R.; Evans, A.; Luessi, F.; Zipp, F.; Giacomini, P.S.; Antel, J.; Bar-Or, A. Dimethyl fumarate-induced lymphopenia in MS due to differential T-cell subset apoptosis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e340. [Google Scholar] [CrossRef] [Green Version]
- Longbrake, E.E.; Ramsbottom, M.J.; Cantoni, C.; Ghezzi, L.; Cross, A.H.; Piccio, L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult. Scler. 2016, 22, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Klinsing, S.; Posevitz-Fejfar, A.; Wiendl, H.; Klotz, L. Dimethyl fumarate treatment alters circulating T helper cell subsets in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e183. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wang, Q.; Mao, G.; Dowling, C.A.; Lundy, S.K.; Mao-Draayer, Y. Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J. Immunol. 2017, 198, 3069–3080. [Google Scholar] [CrossRef] [Green Version]
- Medina, S.; Villarrubia, N.; Sainz de la Maza, S.; Lifante, J.; Costa-Frossard, L.; Roldan, E.; Picon, C.; Alvarez-Cermeno, J.C.; Villar, L.M. Optimal response to dimethyl fumarate associates in MS with a shift from an inflammatory to a tolerogenic blood cell profile. Mult. Scler. 2018, 24, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Polman, C.; Pozzilli, C.; Thompson, A.; Beckmann, K.; Dahlke, F. Final analysis of the european multicenter trial on IFNbeta-1b in secondary-progressive MS. Neurology 2001, 57, 1969–1975. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Schwab, S.R.; Cyster, J.G. Finding a way out: Lymphocyte egress from lymphoid organs. Nat. Immunol. 2007, 8, 1295–1301. [Google Scholar] [CrossRef]
- Pelletier, D.; Hafler, D.A. Fingolimod for multiple sclerosis. N. Engl. J. Med. 2012, 366, 339–347. [Google Scholar] [CrossRef]
- Mehling, M.; Brinkmann, V.; Antel, J.; Bar-Or, A.; Goebels, N.; Vedrine, C.; Kristofic, C.; Kuhle, J.; Lindberg, R.L.; Kappos, L. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology 2008, 71, 1261–1267. [Google Scholar] [CrossRef]
- Serpero, L.D.; Filaci, G.; Parodi, A.; Battaglia, F.; Kalli, F.; Brogi, D.; Mancardi, G.L.; Uccelli, A.; Fenoglio, D. Fingolimod modulates peripheral effector and regulatory T cells in MS patients. J. Neuroimmune. Pharmacol. 2013, 8, 1106–1113. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Villar, M.; Raddassi, K.; Danielsen, A.C.; Guarnaccia, J.; Hafler, D.A. Fingolimod modulates T cell phenotype and regulatory T cell plasticity in vivo. J. Autoimmun. 2019, 96, 40–49. [Google Scholar] [CrossRef]
- Engelhardt, B.; Kappos, L. Natalizumab: Targeting alpha4-integrins in multiple sclerosis. Neurodegener. Dis. 2008, 5, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Stuve, O.; Bennett, J.L. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (tysabri) in inflammatory diseases. CNS Drug Rev. 2007, 13, 79–95. [Google Scholar] [CrossRef]
- Kivisakk, P.; Healy, B.C.; Viglietta, V.; Quintana, F.J.; Hootstein, M.A.; Weiner, H.L.; Khoury, S.J. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology 2009, 72, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Coisne, C.; Mao, W.; Engelhardt, B. Cutting edge: Natalizumab blocks adhesion but not initial contact of human T cells to the blood-brain barrier in vivo in an animal model of multiple sclerosis. J. Immunol. 2009, 182, 5909–5913. [Google Scholar] [CrossRef] [Green Version]
- Berger, B.; Baumgartner, A.; Rauer, S.; Mader, I.; Luetzen, N.; Farenkopf, U.; Stich, O. Severe disease reactivation in four patients with relapsing-remitting multiple sclerosis after fingolimod cessation. J. Neuroimmunol. 2015, 282, 118–122. [Google Scholar] [CrossRef]
- Hatcher, S.E.; Waubant, E.; Nourbakhsh, B.; Crabtree-Hartman, E.; Graves, J.S. Rebound syndrome in patients with multiple sclerosis after cessation of fingolimod treatment. JAMA Neurol. 2016, 73, 790–794. [Google Scholar] [CrossRef] [Green Version]
- Gueguen, A.; Roux, P.; Deschamps, R.; Moulignier, A.; Bensa, C.; Savatovsky, J.; Heran, F.; Gout, O. Abnormal inflammatory activity returns after natalizumab cessation in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1038–1040. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Koch-Henriksen, N.; Petersen, T.; Ravnborg, M.; Oturai, A.; Sellebjerg, F. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy in highly active MS patients. J. Neurol. 2014, 261, 1170–1177. [Google Scholar] [CrossRef]
- Campagnolo, D.; Dong, Q.; Lee, L.; Ho, P.R.; Amarante, D.; Koendgen, H. Statistical analysis of pml incidences of natalizumab-treated patients from 2009 to 2016: Outcomes after introduction of the stratify JCV(r) dxselect antibody assay. J. Neurovirol. 2016, 22, 880–881. [Google Scholar] [CrossRef] [Green Version]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in multiple sclerosis: Mechanism of action and beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef]
- Milo, R. The efficacy and safety of daclizumab and its potential role in the treatment of multiple sclerosis. Ther. Adv. Neurol. Disord. 2014, 7, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Wiendl, H.; Gross, C.C. Modulation of il-2ralpha with daclizumab for treatment of multiple sclerosis. Nat. Rev. Neurol. 2013, 9, 394–404. [Google Scholar] [CrossRef]
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/zinbryta (accessed on 18 May 2018).
- European Medicines Agency Home Page. Available online: https://www.ema.europa.eu/en/medicines/human/referrals/lemtrada (accessed on 19 January 2020).
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Gingele, S.; Jacobus, T.L.; Konen, F.F.; Hummert, M.W.; Suhs, K.W.; Schwenkenbecher, P.; Ahlbrecht, J.; Mohn, N.; Muschen, L.H.; Bonig, L.; et al. Ocrelizumab depletes CD20(+) T cells in multiple sclerosis patients. Cells 2018, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Holley, J.E.; Bremer, E.; Kendall, A.C.; de Bruyn, M.; Helfrich, W.; Tarr, J.M.; Newcombe, J.; Gutowski, N.J.; Eggleton, P. CD20+ inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Mult. Scler. Relat. Disord. 2014, 3, 650–658. [Google Scholar] [CrossRef]
- Bittner, S.; Wiendl, H. Neuroimmunotherapies targeting T cells: From pathophysiology to therapeutic applications. Neurotherapeutics 2016, 13, 4–19. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; van der Heijde, D.; Landewe, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17a monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, O.; Alvarez-Cermeno, J.C.; Arnal-Garcia, C.; Arroyo-Gonzalez, R.; Brieva, L.; Calles-Hernandez, M.C.; Casanova-Estruch, B.; Comabella, M.; Garcia-Merino, J.A.; Izquierdo, G.; et al. Review of the novelties presented at the 29th congress of the european committee for treatment and research in multiple sclerosis (ECTRIMS) (iii). Rev. Neurol. 2014, 59, 371–379. [Google Scholar]
- van der Heijde, D.; Cheng-Chung Wei, J.; Dougados, M.; Mease, P.; Deodhar, A.; Maksymowych, W.P.; Van den Bosch, F.; Sieper, J.; Tomita, T.; Landewe, R.; et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet 2018, 392, 2441–2451. [Google Scholar]
- van der Heijde, D.; Gladman, D.D.; Kishimoto, M.; Okada, M.; Rathmann, S.S.; Moriarty, S.R.; Shuler, C.L.; Carlier, H.; Benichou, O.; Mease, P.J. Efficacy and safety of ixekizumab in patients with active psoriatic arthritis: 52-week results from a phase iii study (SPIRIT-P1). J. Rheumatol. 2018, 45, 367–377. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells 2020, 9, 482. https://doi.org/10.3390/cells9020482
Kunkl M, Frascolla S, Amormino C, Volpe E, Tuosto L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells. 2020; 9(2):482. https://doi.org/10.3390/cells9020482
Chicago/Turabian StyleKunkl, Martina, Simone Frascolla, Carola Amormino, Elisabetta Volpe, and Loretta Tuosto. 2020. "T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis" Cells 9, no. 2: 482. https://doi.org/10.3390/cells9020482
APA StyleKunkl, M., Frascolla, S., Amormino, C., Volpe, E., & Tuosto, L. (2020). T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells, 9(2), 482. https://doi.org/10.3390/cells9020482