Dynamic Signatures of the Epigenome: Friend or Foe?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Molecular Mechanisms behind the Inheritance of DNA Methylation and Posttranslational Histone Modifications
3. Germline Inheritance of Epimutation
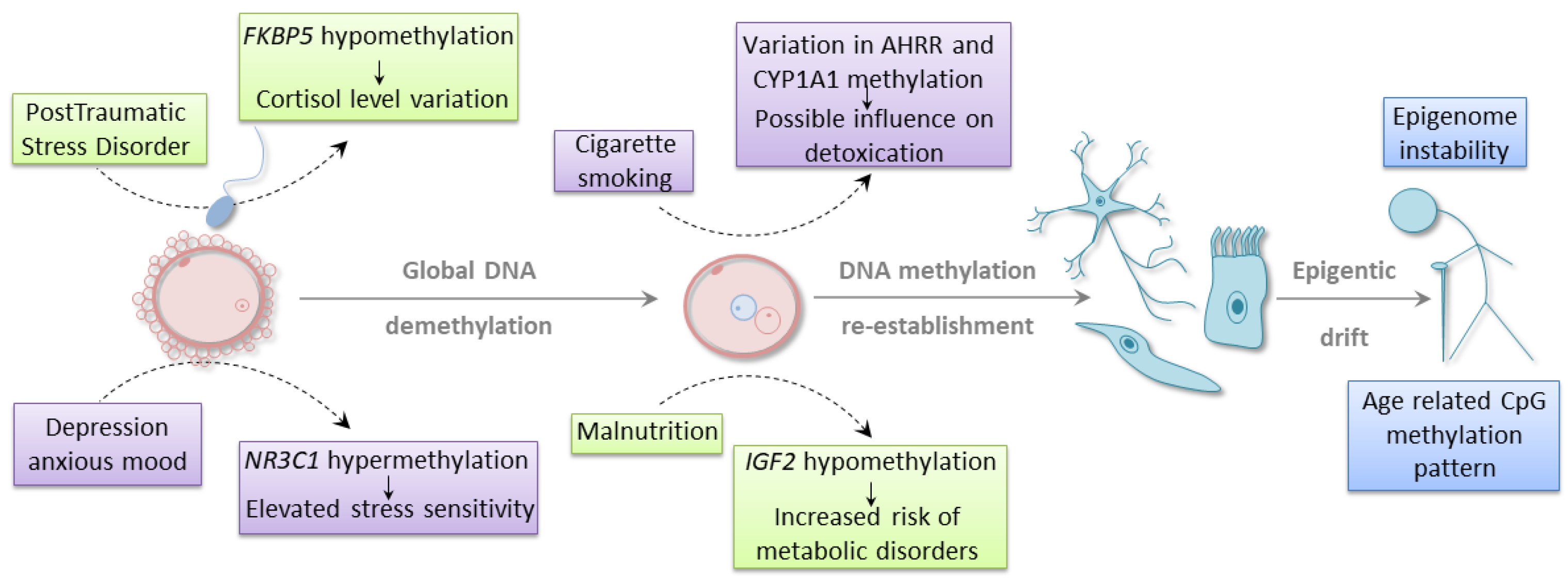
4. Environmental Influence on Epigenetics
5. Epigenetic Signature in Species Evolution
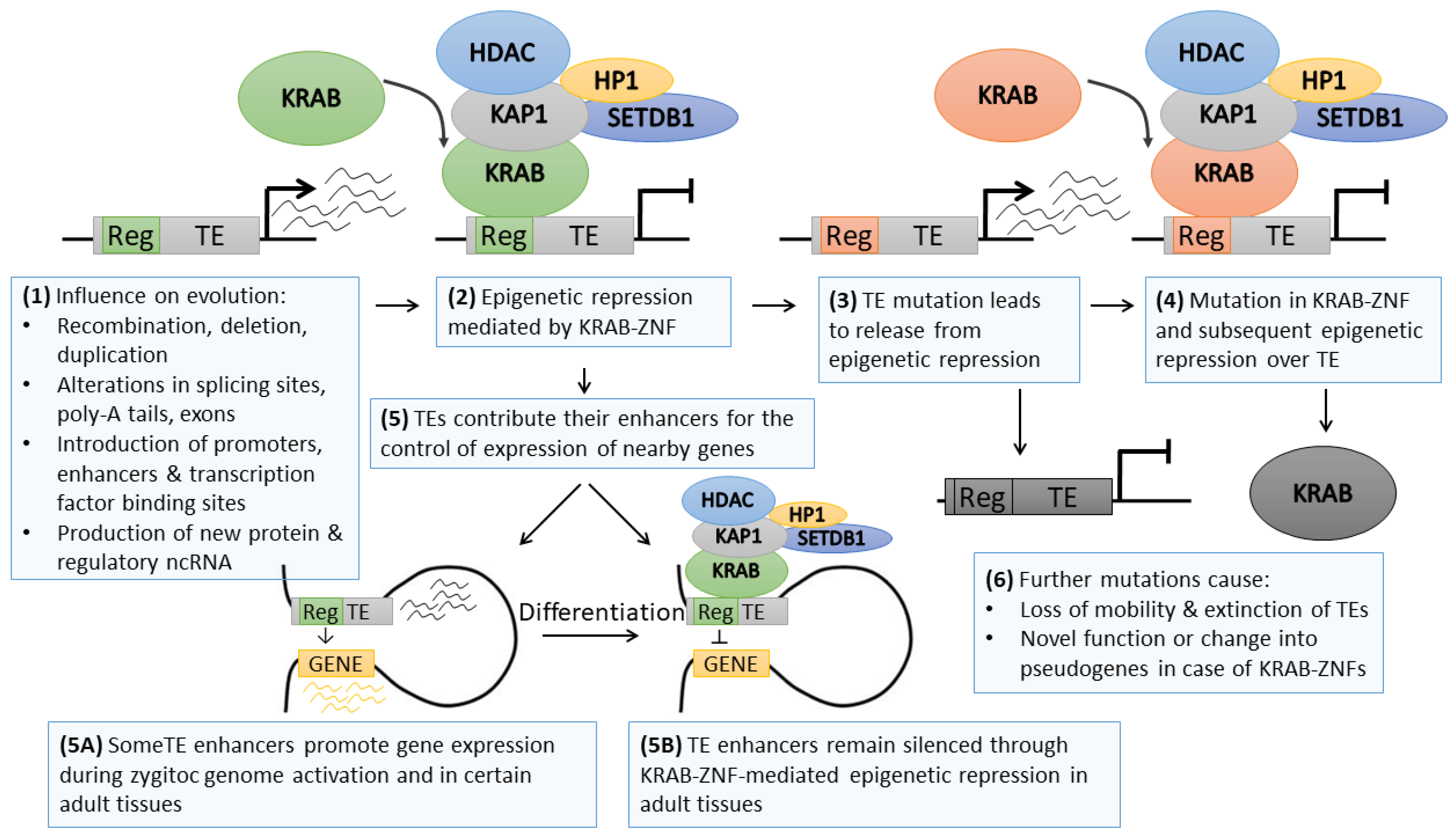
6. The Co-Evolution between Transposable Elements and their Epigenetic Repressors
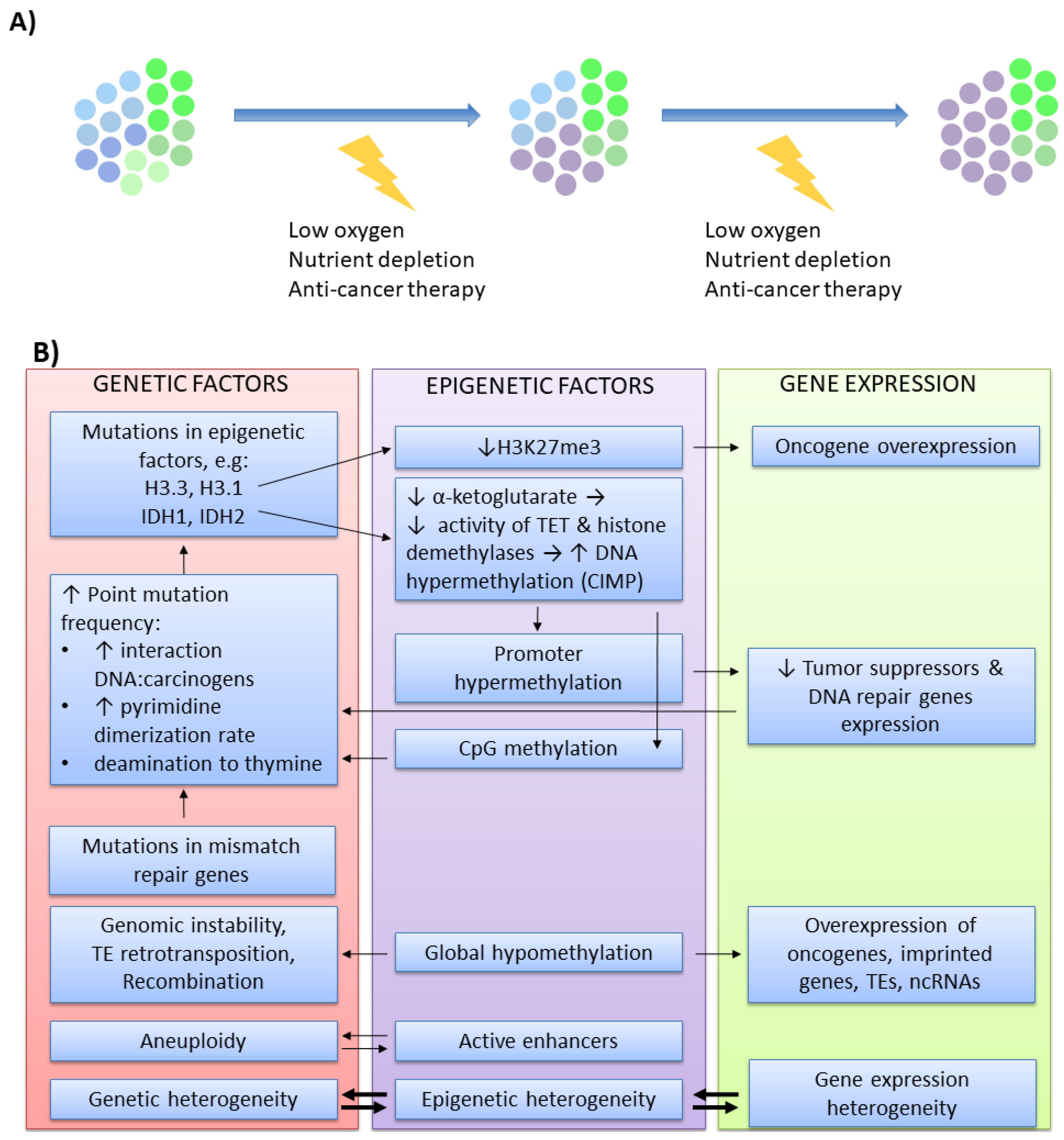
7. Cancer Microevolution—Focus on Epigenomic Alterations
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Giuliani, C.; Bacalini, M.G.; Sazzini, M.; Pirazzini, C.; Franceschi, C.; Garagnani, P.; Luiselli, D. The epigenetic side of human adaptation: Hypotheses, evidences and theories. Ann. Hum. Biol. 2015, 42, 1–9. [Google Scholar] [CrossRef]
- Tollefsbol, T.O. An overview of epigenetics. In Handbook of Epigenetics. The New Molecular and Medical Genetics, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 3–8. [Google Scholar]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabin, J.; Fortuny, A.; Polo, S.E. Epigenome Maintenance in Response to DNA Damage. Mol. Cell 2016, 62, 712–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. The Strategy of the Genes; George Allen & Unwin: London, UK, 1957. [Google Scholar]
- Corry, G.N.; Tanasijevic, B.; Barry, E.R.; Krueger, W.; Rasmussen, T.P. Epigenetic regulatory mechanisms during preimplantation development. Birth Defects Res. Part C Embryo Today Rev. 2009, 87, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, E.; Hochedlinger, K. Chromatin dynamics during cellular reprogramming. Nature 2013, 502, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladych, M.; Andrzejewska, A.; Oleksiewicz, U.; Estecio, M.R. Epigenetic mechanisms of induced pluripotency. Contemp. Oncol. (Pozn. Pol.) 2015, 19, A30–A38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Li, C.; Peng, X.; Zhou, Z.; Weinstein, J.N.; Liang, H. A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 2018, 173, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Kara, N.; Cherry, A.; Sinha, A.U.; Zhu, N.; Bernt, K.M.; Cahan, P.; Marcarci, B.O.; Unternaehrer, J.; Gupta, P.B.; et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 2012, 483, 598–602. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Alajem, A.; Meshorer, E.; Ramalho-Santos, M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2011, 12, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lempradl, A. Germ cell-mediated mechanisms of epigenetic inheritance. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sarkies, P. Molecular mechanisms of epigenetic inheritance: Possible evolutionary implications. Semin. Cell Dev. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Lambrot, R.; Xu, C.; Saint-Phar, S.; Chountalos, G.; Cohen, T.; Paquet, M.; Suderman, M.; Hallett, M.; Kimmins, S. Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Commun. 2013, 4, 2889. [Google Scholar] [CrossRef]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A.C.; Fung, E.; Bieda, M.C.; Snyder, F.F.; Gravel, R.A.; Cross, J.C.; et al. Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 2013, 155, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Pogribny, I.P.; Tryndyak, V.P.; Muskhelishvili, L.; Rusyn, I.; Ross, S.A. Methyl Deficiency, Alterations in Global Histone Modifications, and Carcinogenesis. J. Nutr. 2007, 137, 216S–222S. [Google Scholar] [CrossRef] [Green Version]
- Siklenka, K.; Erkek, S.; Godmann, M.; Lambrot, R.; McGraw, S.; Lafleur, C.; Cohen, T.; Xia, J.; Suderman, M.; Hallett, M.; et al. Disruption of histone methylation in developing sperm impairs offspring health transgenerationally. Science 2015, 350, aab2006. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loda, A.; Brandsma, J.H.; Vassilev, I.; Servant, N.; Loos, F.; Amirnasr, A.; Splinter, E.; Barillot, E.; Poot, R.A.; Heard, E.; et al. Genetic and epigenetic features direct differential efficiency of Xist-mediated silencing at X-chromosomal and autosomal locations. Nat. Commun. 2017, 8, 690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohm, V.; Hieb, A.R.; Andrews, A.J.; Gansen, A.; Rocker, A.; Toth, K.; Luger, K.; Langowski, J. Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res. 2011, 39, 3093–3102. [Google Scholar] [CrossRef]
- Xu, M.; Long, C.; Chen, X.; Huang, C.; Chen, S.; Zhu, B. Partitioning of Histone H3-H4 Tetramers During DNA Replication–Dependent Chromatin Assembly. Science 2010, 328, 94. [Google Scholar] [CrossRef] [Green Version]
- Alabert, C.; Barth, T.K.; Reveron-Gomez, N.; Sidoli, S.; Schmidt, A.; Jensen, O.N.; Imhof, A.; Groth, A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015, 29, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef]
- Ramachandran, S.; Henikoff, S. Replicating Nucleosomes. Sci. Adv. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Escobar, T.M.; Oksuz, O.; Saldana-Meyer, R.; Descostes, N.; Bonasio, R.; Reinberg, D. Active and Repressed Chromatin Domains Exhibit Distinct Nucleosome Segregation during DNA Replication. Cell 2019, 179, 953–963. [Google Scholar] [CrossRef]
- Moazed, D. Mechanisms for the inheritance of chromatin states. Cell 2011, 146, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Reveron-Gomez, N.; Gonzalez-Aguilera, C.; Stewart-Morgan, K.R.; Petryk, N.; Flury, V.; Graziano, S.; Johansen, J.V.; Jakobsen, J.S.; Alabert, C.; Groth, A. Accurate Recycling of Parental Histones Reproduces the Histone Modification Landscape during DNA Replication. Mol. Cell 2018, 72, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando-Herraez, I.; Heyn, H.; Fernandez-Callejo, M.; Vidal, E.; Fernandez-Bellon, H.; Prado-Martinez, J.; Sharp, A.J.; Esteller, M.; Marques-Bonet, T. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 2015, 43, 8204–8214. [Google Scholar] [CrossRef] [PubMed]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leseva, M.N.; Grand, R.J.; Klett, H.; Boerries, M.; Busch, H.; Binder, A.M.; Michels, K.B. Differences in DNA Methylation and Functional Expression in Lactase Persistent and Non-persistent Individuals. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R.; et al. Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metab. 2015, 21, 905–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, H.J.; Vieux, K.F. Epigenetic inheritance through the female germ-line: The known, the unknown, and the possible. Semin. Cell Dev. Biol. 2015, 43, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.H.; Sauria, M.E.G.; Lyu, X.; Cheema, M.S.; Ausio, J.; Taylor, J.; Corces, V.G. Chromatin States in Mouse Sperm Correlate with Embryonic and Adult Regulatory Landscapes. Cell Rep. 2017, 18, 1366–1382. [Google Scholar] [CrossRef]
- van de Werken, C.; van der Heijden, G.W.; Eleveld, C.; Teeuwssen, M.; Albert, M.; Baarends, W.M.; Laven, J.S.; Peters, A.H.; Baart, E.B. Paternal heterochromatin formation in human embryos is H3K9/HP1 directed and primed by sperm-derived histone modifications. Nat. Commun. 2014, 5, 5868. [Google Scholar] [CrossRef] [Green Version]
- Larriba, E.; del Mazo, J. Role of Non-Coding RNAs in the Transgenerational Epigenetic Transmission of the Effects of Reprotoxicants. Int. J. Mol. Sci. 2016, 17, 452. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.H.; Peng, H.; Zhang, X.; Zhang, Y.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, M.; Moore, G.E. The role of imprinted genes in humans. Mol. Asp. Med. 2013, 34, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Soubry, A.; Murphy, S.K.; Wang, F.; Huang, Z.; Vidal, A.C.; Fuemmeler, B.F.; Kurtzberg, J.; Murtha, A.; Jirtle, R.L.; Schildkraut, J.M.; et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. (Lond.) 2015, 39, 650–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soubry, A.; Schildkraut, J.M.; Murtha, A.; Wang, F.; Huang, Z.; Bernal, A.; Kurtzberg, J.; Jirtle, R.L.; Murphy, S.K.; Hoyo, C. Paternal obesity is associated with IGF2 hypomethylation in newborns: Results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. 2013, 11, 29. [Google Scholar] [CrossRef] [Green Version]
- van der Graaf, A.; Wardenaar, R.; Neumann, D.A.; Taudt, A.; Shaw, R.G.; Jansen, R.C.; Schmitz, R.J.; Colome-Tatche, M.; Johannes, F. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. USA 2015, 112, 6676–6681. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; West, J.; Beck, S. Age-associated epigenetic drift: Implications, and a case of epigenetic thrift? Hum. Mol. Genet. 2013, 22, R7–R15. [Google Scholar] [CrossRef]
- Joubert, B.R.; Håberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Huang, Z.; Hoyo, C.; Midttun, Ø.; Cupul-Uicab, L.A.; et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1431. [Google Scholar] [CrossRef] [Green Version]
- Monick, M.M.; Beach, S.R.H.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Hompes, T.; Izzi, B.; Gellens, E.; Morreels, M.; Fieuws, S.; Pexsters, A.; Schops, G.; Dom, M.; Van Bree, R.; Freson, K.; et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J. Psychiatry Res. 2013, 47, 880–891. [Google Scholar] [CrossRef]
- Oberlander, T.F.; Weinberg, J.; Papsdorf, M.; Grunau, R.; Misri, S.; Devlin, A.M. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008, 3, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehuda, R.; Daskalakis, N.P.; Bierer, L.M.; Bader, H.N.; Klengel, T.; Holsboer, F.; Binder, E.B. Holocaust Exposure Induced Intergenerational Effects on FKBP5 Methylation. Biol. Psychiatry 2016, 80, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Lumey, L.H.; Stein, A.D.; Susser, E. Prenatal famine and adult health. Annu. Rev. Public Health 2011, 32, 237–262. [Google Scholar] [CrossRef] [Green Version]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Dias, B.G.; Ressler, K.J. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 2014, 17, 89–96. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Yang, C.R.; Wei, Y.P.; Zhao, Z.A.; Hou, Y.; Schatten, H.; Sun, Q.Y. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 1873–1878. [Google Scholar] [CrossRef] [Green Version]
- Labrie, V.; Buske, O.J.; Oh, E.; Jeremian, R.; Ptak, C.; Gasiunas, G.; Maleckas, A.; Petereit, R.; Zvirbliene, A.; Adamonis, K.; et al. Lactase nonpersistence is directed by DNA-variation-dependent epigenetic aging. Nat. Struct. Mol. Biol. 2016, 23, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Alkorta-Aranburu, G.; Beall, C.M.; Witonsky, D.B.; Gebremedhin, A.; Pritchard, J.K.; Di Rienzo, A. The genetic architecture of adaptations to high altitude in Ethiopia. PLoS Genet. 2012, 8, e1003110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendizabal, I.; Keller, T.E.; Zeng, J.; Yi, S.V. Epigenetics and evolution. Integr. Comp. Biol. 2014, 54, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, A.A.; Bell, J.T.; Marioni, J.C.; Pritchard, J.K.; Gilad, Y. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011, 7, e1001316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Konopka, G.; Hunt, B.G.; Preuss, T.M.; Geschwind, D.; Yi, S.V. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am. J. Hum. Genet. 2012, 91, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernando-Herraez, I.; Prado-Martinez, J.; Garg, P.; Fernandez-Callejo, M.; Heyn, H.; Hvilsom, C.; Navarro, A.; Esteller, M.; Sharp, A.J.; Marques-Bonet, T. Dynamics of DNA methylation in recent human and great ape evolution. PLoS Genet. 2013, 9, e1003763. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Pääbo, S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010, 38, e87. [Google Scholar] [CrossRef] [Green Version]
- Gokhman, D.; Lavi, E.; Prufer, K.; Fraga, M.F.; Riancho, J.A.; Kelso, J.; Paabo, S.; Meshorer, E.; Carmel, L. Reconstructing the DNA Methylation Maps of the Neandertal and the Denisovan. Science 2014, 344, 523–527. [Google Scholar] [CrossRef]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Carja, O.; MacIsaac, J.L.; Mah, S.M.; Henn, B.M.; Kobor, M.S.; Feldman, M.W.; Fraser, H.B. Worldwide patterns of human epigenetic variation. Nat. Ecol. Evol. 2017, 1, 1577–1583. [Google Scholar] [CrossRef]
- Scott, A.F.; Schmeckpeper, B.J.; Abdelrazik, M.; Comey, C.T.; O′Hara, B.; Rossiter, J.P.; Cooley, T.; Heath, P.; Smith, K.D.; Margolet, L. Origin of the human L1 elements: Proposed progenitor genes deduced from a consensus DNA sequence. Genomics 1987, 1, 113–125. [Google Scholar] [CrossRef]
- Belshaw, R.; Pereira, V.; Katzourakis, A.; Talbot, G.; Paces, J.; Burt, A.; Tristem, M. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2004, 101, 4894–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedli, M.; Trono, D. The developmental control of transposable elements and the evolution of higher species. Annu. Rev. Cell Dev. Biol. 2015, 31, 429–451. [Google Scholar] [CrossRef]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Kapusta, A.; Kronenberg, Z.; Lynch, V.J.; Zhuo, X.; Ramsay, L.; Bourque, G.; Yandell, M.; Feschotte, C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet. 2013, 9, e1003470. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Glazko, G.V.; Koonin, E.V. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. TIG 2003, 19, 68–72. [Google Scholar] [CrossRef]
- Bejerano, G.; Lowe, C.B.; Ahituv, N.; King, B.; Siepel, A.; Salama, S.R.; Rubin, E.M.; Kent, W.J.; Haussler, D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature 2006, 441, 87–90. [Google Scholar] [CrossRef]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Goncalves, A.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunarso, G.; Chia, N.Y.; Jeyakani, J.; Hwang, C.; Lu, X.; Chan, Y.S.; Ng, H.H.; Bourque, G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010, 42, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Pontis, J.; Planet, E.; Offner, S.; Turelli, P.; Duc, J.; Coudray, A.; Theunissen, T.W.; Jaenisch, R.; Trono, D. Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 2019, 24, 724–735.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trono, D. Transposable Elements, Polydactyl Proteins, and the Genesis of Human-Specific Transcription Networks. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Mommert, M.; Tabone, O.; Oriol, G.; Cerrato, E.; Guichard, A.; Naville, M.; Fournier, P.; Volff, J.N.; Pachot, A.; Monneret, G.; et al. LTR-retrotransposon transcriptome modulation in response to endotoxin-induced stress in PBMCs. BMC Genom. 2018, 19, 522. [Google Scholar] [CrossRef]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [Green Version]
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Rowe, H.M.; Trono, D. Dynamic control of endogenous retroviruses during development. Virology 2011, 411, 273–287. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sripathy, S.P.; Stevens, J.; Schultz, D.C. The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell Biol. 2006, 26, 8623–8638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Diaz, N.; Ecco, G.; Coluccio, A.; Kapopoulou, A.; Yazdanpanah, B.; Friedli, M.; Duc, J.; Jang, S.M.; Turelli, P.; Trono, D. Evolutionally dynamic L1 regulation in embryonic stem cells. Genes Dev. 2014, 28, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Castro-Diaz, N.; Marzetta, F.; Kapopoulou, A.; Raclot, C.; Duc, J.; Tieng, V.; Quenneville, S.; Trono, D. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res. 2014, 24, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Brattas, P.L.; Jonsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, Y.; Macfarlan, T.S. The Role of KRAB-ZFPs in Transposable Element Repression and Mammalian Evolution. Trends Genet. TIG 2017, 33, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development (Camb. Engl.) 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jonsson, M.E.; Manne, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Coluccio, A.; Thorball, C.W.; Planet, E.; Shi, H.; Offner, S.; Turelli, P.; Imbeault, M.; Ferguson-Smith, A.C.; Trono, D. ZNF445 is a primary regulator of genomic imprinting. Genes Dev. 2019, 33, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, Y.; Hoang, D.; Tinkham, M.; Patel, A.; Sun, M.A.; Wolf, G.; Baker, M.; Chien, H.C.; Lai, K.N.; et al. A placental growth factor is silenced in mouse embryos by the zinc finger protein ZFP568. Science 2017, 356, 757–759. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Issa, J.J. Introduction: Cancer as an Epigenetic Disease. Cancer J. (Sudbury Mass) 2017, 23, 255–256. [Google Scholar] [CrossRef]
- Brocato, J.; Costa, M. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit. Rev. Toxicol. 2013, 43, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Hitchins, M.P. Inheritance of epigenetic aberrations (constitutional epimutations) in cancer susceptibility. Adv. Genet. 2010, 70, 201–243. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugué, P.-A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C.; et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat. Commun. 2018, 9, 867. [Google Scholar] [CrossRef] [PubMed]
- Quinonez-Silva, G.; Davalos-Salas, M.; Recillas-Targa, F.; Ostrosky-Wegman, P.; Aranda, D.A.; Benitez-Bribiesca, L. Monoallelic germline methylation and sequence variant in the promoter of the RB1 gene: A possible constitutive epimutation in hereditary retinoblastoma. Clin. Epigenet. 2016, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.H.; Waterland, R.A.; Zhang, P.; Schady, D.; Chen, M.H.; Guan, Y.; Gadkari, M.; Shen, L. Targeted p16(Ink4a) epimutation causes tumorigenesis and reduces survival in mice. J. Clin. Investig. 2014, 124, 3708–3712. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Greger, V.; Passarge, E.; Hopping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Huang, S.; Gulzar, Z.G.; Salari, K.; Lapointe, J.; Brooks, J.D.; Pollack, J.R. Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene 2012, 31, 4164–4170. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Mayes, K.; Qiu, Z.; Alhazmi, A.; Landry, J.W. ATP-dependent chromatin remodeling complexes as novel targets for cancer therapy. Adv. Cancer Res. 2014, 121, 183–233. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xiao, Y.; Wang, W.; Wang, Q.; Yearsley, K.; Wani, A.A.; Yan, Q.; Gao, J.X.; Shetuni, B.S.; Barsky, S.H. Inhibition of expression of the chromatin remodeling gene, SNF2L, selectively leads to DNA damage, growth inhibition, and cancer cell death. Mol. Cancer Res. 2009, 7, 1984–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, A.; Niu, J.; Qu, Q.; Zhao, N.; Ruan, Y.; Nadas, A.; Chervona, Y.; Wu, F.; Sun, H.; Hayes, R.B.; et al. Global levels of histone modifications in peripheral blood mononuclear cells of subjects with exposure to nickel. Environ. Health Perspect. 2012, 120, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Kluz, T.; Harder, M.E.; Xiong, J.; Costa, M. Heterochromatinization as a potential mechanism of nickel-induced carcinogenesis. Biochemistry 2009, 48, 4626–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, S.; Dasgupta, U.B.; Guhamazumder, D.; Gupta, M.; Chaudhuri, U.; Lahiri, S.; Das, S.; Ghosh, N.; Chatterjee, D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. Off. J. Soc. Toxicol. 2006, 89, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.J.; Novak, P.; Eblin, K.E.; Gandolfi, A.J.; Futscher, B.W. Epigenetic remodeling during arsenical-induced malignant transformation. Carcinogenesis 2008, 29, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Benbrahim-Tallaa, L.; Waterland, R.A.; Dill, A.L.; Webber, M.M.; Waalkes, M.P. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ. Health Perspect. 2007, 115, 1454–1459. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Xu, L.; Song, S.; Zhu, C.; Wu, Q.; Zhang, L.; Wu, L. Effects of long-term low-dose cadmium exposure on genomic DNA methylation in human embryo lung fibroblast cells. Toxicology 2008, 244, 49–55. [Google Scholar] [CrossRef]
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Wong, J.J.; Suthers, G.; Suter, C.M.; Martin, D.I.; Hawkins, N.J.; Ward, R.L. Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med. 2007, 356, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morak, M.; Schackert, H.K.; Rahner, N.; Betz, B.; Ebert, M.; Walldorf, C.; Royer-Pokora, B.; Schulmann, K.; von Knebel-Doeberitz, M.; Dietmaier, W.; et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur. J. Hum. Genet. EJHG 2008, 16, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raval, A.; Tanner, S.M.; Byrd, J.C.; Angerman, E.B.; Perko, J.D.; Chen, S.S.; Hackanson, B.; Grever, M.R.; Lucas, D.M.; Matkovic, J.J.; et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 2007, 129, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell, C.; Krypuy, M.; Wong, E.M.; Loughrey, M.B.; Dobrovic, A. BRCA1 promoter methylation in peripheral blood DNA of mutation negative familial breast cancer patients with a BRCA1 tumour phenotype. Breast Cancer Res. BCR 2008, 10, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- TCGA, N. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- TCGA, N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Zheng, J.; Huang, X.; Tan, W.; Yu, D.; Du, Z.; Chang, J.; Wei, L.; Han, Y.; Wang, C.; Che, X.; et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016, 48, 747–757. [Google Scholar] [CrossRef]
- Sheaffer, K.L.; Elliott, E.N.; Kaestner, K.H. DNA Hypomethylation Contributes to Genomic Instability and Intestinal Cancer Initiation. Cancer Prev. Res. (Phila. PA) 2016, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Franke, G.; Bausch, B.; Hoffmann, M.M.; Cybulla, M.; Wilhelm, C.; Kohlhase, J.; Scherer, G.; Neumann, H.P. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: Molecular characterization and genotype-phenotype correlations in VHL patients. Hum. Mutat. 2009, 30, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J.; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Klift, H.; Wijnen, J.; Wagner, A.; Verkuilen, P.; Tops, C.; Otway, R.; Kohonen-Corish, M.; Vasen, H.; Oliani, C.; Barana, D.; et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 2005, 44, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Risques, R.A.; Toyota, M.; Capella, G.; Moreno, V.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res. 2001, 61, 4689–4692. [Google Scholar]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.P.; Markowitz, S.; Willson, J.K.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [Green Version]
- You, Y.H.; Pfeifer, G.P. Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine residues in solar UV mutagenesis. J. Mol. Biol. 2001, 305, 389–399. [Google Scholar] [CrossRef]
- Oakes, C.C.; Claus, R.; Gu, L.; Assenov, Y.; Hullein, J.; Zucknick, M.; Bieg, M.; Brocks, D.; Bogatyrova, O.; Schmidt, C.R.; et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014, 4, 348–361. [Google Scholar] [CrossRef] [Green Version]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, Y.; Straussman, R.; Keshet, I.; Farkash, S.; Hecht, M.; Zimmerman, J.; Eden, E.; Yakhini, Z.; Ben-Shushan, E.; Reubinoff, B.E.; et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet. 2007, 39, 232–236. [Google Scholar] [CrossRef]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Court, F.; Arnaud, P. An annotated list of bivalent chromatin regions in human ES cells: A new tool for cancer epigenetic research. Oncotarget 2017, 8, 4110–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, L.; Huang, W.; Bellani, M.; Seidman, M.M.; Wu, K.; Fan, D.; Nie, Y.; Cai, Y.; Zhang, Y.W.; Yu, L.R.; et al. CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer Cell 2017, 31, 653–668 e657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Bhutani, M.; Pathak, A.K.; Lang, W.; Ren, H.; Jelinek, J.; He, R.; Shen, L.; Issa, J.P.; Mao, L. Delta DNMT3B variants regulate DNA methylation in a promoter-specific manner. Cancer Res. 2007, 67, 10647–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cylwa, R.; Kielczewski, K.; Machnik, M.; Oleksiewicz, U.; Biecek, P. KRAB ZNF explorer -the online tool for the exploration of the transcriptomic profiles of KRAB-ZNF factors in The Cancer Genome Atlas. Bioinformatics (Oxf. Engl.) 2019. [Google Scholar] [CrossRef] [PubMed]
- Machnik, M.; Cylwa, R.; Kielczewski, K.; Biecek, P.; Liloglou, T.; Mackiewicz, A.; Oleksiewicz, U. The expression signature of cancer-associated KRAB-ZNF factors identified in TCGA pan-cancer transcriptomic data. Mol. Oncol. 2019, 13, 701–724. [Google Scholar] [CrossRef] [Green Version]
- Oleksiewicz, U.; Gladych, M.; Raman, A.T.; Heyn, H.; Mereu, E.; Chlebanowska, P.; Andrzejewska, A.; Sozanska, B.; Samant, N.; Fak, K.; et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Rep. 2017, 9, 2065–2080. [Google Scholar] [CrossRef] [Green Version]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D′Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63. [Google Scholar] [CrossRef]
- Shahrzad, S.; Bertrand, K.; Minhas, K.; Coomber, B. Induction of DNA Hypomethylation by Tumor Hypoxia. Epigenetics 2007, 2, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Camuzi, D.; de Amorim, I.S.S.; Ribeiro Pinto, L.F.; Oliveira Trivilin, L.; Mencalha, A.L.; Soares Lima, S.C. Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells 2019, 8, 300. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-stabilized EPAS1 proteins transactivate DNMT1 and cause promoter hypermethylation and transcription inhibition of EPAS1 in non-small cell lung cancer. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Wylie, B.; Chee, J.; Forbes, C.A.; Booth, M.; Stone, S.R.; Buzzai, A.; Abad, A.; Foley, B.; Cruickshank, M.N.; Waithman, J. Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. Oncoimmunology 2019, 8, 1609874. [Google Scholar] [CrossRef] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergstrom, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sonmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machnik, M.; Oleksiewicz, U. Dynamic Signatures of the Epigenome: Friend or Foe? Cells 2020, 9, 653. https://doi.org/10.3390/cells9030653
Machnik M, Oleksiewicz U. Dynamic Signatures of the Epigenome: Friend or Foe? Cells. 2020; 9(3):653. https://doi.org/10.3390/cells9030653
Chicago/Turabian StyleMachnik, Marta, and Urszula Oleksiewicz. 2020. "Dynamic Signatures of the Epigenome: Friend or Foe?" Cells 9, no. 3: 653. https://doi.org/10.3390/cells9030653
APA StyleMachnik, M., & Oleksiewicz, U. (2020). Dynamic Signatures of the Epigenome: Friend or Foe? Cells, 9(3), 653. https://doi.org/10.3390/cells9030653