Total mRNA Quantification in Single Cells: Sarcoma Cell Heterogeneity

 , , ,
, , ,
Abstract
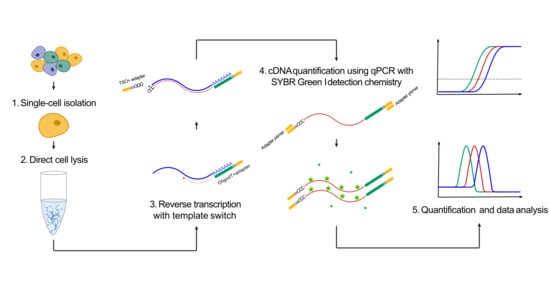
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Total RNA Extraction
2.3. Single-Cell Collection
2.4. Total Polyadenylated RNA Analysis
3. Results
3.1. Development of a Method to Quantify the Polyadenylated Transcriptome of Single Cells
3.2. Individual Sarcoma Cells Reveal Heterogeneity in Total Polyadenylated Transcriptome Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kubista, M.; Dreyer-Lamm, J.; Stahlberg, A. The secrets of the cell. Mol. Asp. Med. 2018, 59, 1–4. [Google Scholar] [CrossRef]
- Bengtsson, M.; Ståhlberg, A.; Rorsman, P.; Kubista, M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [Green Version]
- Hedlund, E.; Deng, Q. Single-cell RNA sequencing: Technical advancements and biological applications. Mol. Asp. Med. 2018, 59, 36–46. [Google Scholar] [CrossRef]
- Han, X.; Wang, R.; Zhou, Y.; Fei, L.; Sun, H.; Lai, S.; Saadatpour, A.; Zhou, Z.; Chen, H.; Ye, F.; et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018, 172, 1091–1107. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Fraticelli, A.; Wolock, S.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Haseley, N.; Brown, U.; Penaranda, C.; Jijon, H.B.; Trombetta, J.J.; Satija, R.; Shalek, A.K.; Xavier, R.J.; Regev, A.; et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell 2015, 162, 1309–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Coate, J.; Doyle, J.J. Variation in transcriptome size: Are we getting the message? Chromosoma 2014, 124, 27–43. [Google Scholar] [CrossRef]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.-B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [Green Version]
- Marguerat, S.; Bahler, J. Coordinating genome expression with cell size. Trends Genet. 2012, 28, 560–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchison, J. Growth During the Cell Cycle. Adv. Clin. Chem. 2003, 226, 165–258. [Google Scholar]
- Hu, Z.; Chen, K.; Xia, Z.; Chavez, M.; Pal, S.; Seol, J.-H.; Chen, C.-C.; Li, W.; Tyler, J.K. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 2014, 28, 396–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Lovén, J.; Kwok, S.-M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Lovén, J.; Orlando, D.A.; Sigova, A.A.; Lin, C.Y.; Rahl, P.B.; Burge, C.B.; Levens, D.L.; Lee, T.; Young, R.A. Revisiting global gene expression analysis. Cell 2012, 151, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Dolatabadi, S.; Candia, J.; Akrap, N.; Vannas, C.; Tomic, T.T.; Losert, W.; Landberg, G.; Åman, P.; Ståhlberg, A. Cell Cycle and Cell Size Dependent Gene Expression Reveals Distinct Subpopulations at Single-Cell Level. Front. Genet. 2017, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.; Kroneis, T.; Jonasson, E.; Larsson, E.; Ståhlberg, A. Transcriptomic Characterization of the Human Cell Cycle in Individual Unsynchronized Cells. J. Mol. Biol. 2017, 429, 3909–3924. [Google Scholar] [CrossRef]
- Åman, P.; Dolatabadi, S.; Svec, D.; Jonasson, E.; Safavi, S.; Andersson, D.; Grundevik, P.; Thomsen, C.; Ståhlberg, A. Regulatory mechanisms, expression levels and proliferation effects of theFUS-DDIT3fusion oncogene in liposarcoma. J. Pathol. 2016, 238, 689–699. [Google Scholar] [CrossRef]
- Rasheed, S.; Toth, E.M.; Arnstein, P.; Gardner, M.B.; Nelson-Rees, W.A. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 1974, 33, 1027–1033. [Google Scholar] [CrossRef]
- Cavazzana, A.O.; Miser, J.S.; Jefferson, J.; Triche, T.J. Experimental evidence for a neural origin of Ewing’s sarcoma of bone. Am. J. Pathol. 1987, 127, 507–518. [Google Scholar] [PubMed]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length rna-seq from single cells using smart-seq2. Nat. Protocols 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, A.; Rusnakova, V.; Forootan, A.; Andĕrová, M.; Kubista, M. RT-qPCR work-flow for single-cell data analysis. Methods 2013, 59, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kroneis, T.; Jonasson, E.; Andersson, D.; Dolatabadi, S.; Ståhlberg, A. Global preamplification simplifies targeted mRNA quantification. Sci. Rep. 2017, 7, 45219. [Google Scholar] [CrossRef] [PubMed]
- Ståhlberg, A.; Gustafsson, C.K.; Engtröm, K.; Thomsen, C.; Dolatabadi, S.; Jonasson, E.; Li, C.-Y.; Ruff, D.; Chen, S.-M.; Åman, P. Normal and Functional TP53 in Genetically Stable Myxoid/Round Cell Liposarcoma. PLoS ONE 2014, 9, e113110. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Paz, A.C.; Wilky, B.; Johnson, B.; Galoian, K.; Rosenberg, A.; Hu, G.; Tinoco, G.; Bodamer, O.; Trent, J.C. Treatment with a Small Molecule Mutant IDH1 Inhibitor Suppresses Tumorigenic Activity and Decreases Production of the Oncometabolite 2-Hydroxyglutarate in Human Chondrosarcoma Cells. PLoS ONE 2015, 10, e0133813. [Google Scholar] [CrossRef]
- Jiang, L.; Schlesinger, F.; Davis, C.A.; Zhang, Y.; Li, R.; Salit, M.; Gingeras, T.R.; Oliver, B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011, 21, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Vallejos, C.A.; Risso, D.; Scialdone, A.; Dudoit, S.; Marioni, J.C. Normalizing single-cell RNA sequencing data: Challenges and opportunities. Nat. Methods 2017, 14, 565–571. [Google Scholar] [CrossRef]
- Aanes, H.; Winata, C.L.; Moen, L.F.; Østrup, O.; Mathavan, S.; Collas, P.; Rognes, T.; Aleström, P. Normalization of RNA-Sequencing Data from Samples with Varying mRNA Levels. PLoS ONE 2014, 9, e89158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Li, X.; He, J.; Zhou, W.; Song, K.; Guo, Y.; Liu, H.; Guan, Q.; Yan, H.; Wang, X.; et al. Identification and characterization of genes with absolute mRNA abundances changes in tumor cells with varied transcriptome sizes. BMC Genom. 2019, 20, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, B.T.; Landry, J.-R. RNA-Seq—quantitative measurement of expression through massively parallel RNA-sequencing. Methods 2009, 48, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Duff, M.O.; Graveley, B.R.; Carmichael, G.G.; Chen, L.-L. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011, 12, R16. [Google Scholar] [CrossRef] [Green Version]
- Verboom, K.; Everaert, C.; Bolduc, N.; Livak, K.J.; Yigit, N.; Rombaut, D.; Anckaert, J.; Lee, S.; Venø, M.T.; Kjems, J.; et al. SMARTer single cell total RNA sequencing. Nucleic Acids Res. 2019, 47, e93. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef] [Green Version]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef]
- Gudnason, H.; Dufva, M.; Bang, D.D.; Wolff, A. Comparison of multiple DNA dyes for real-time PCR: Effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007, 35, e127. [Google Scholar] [CrossRef]
- Ståhlberg, A.; Håkansson, J.; Xian, X.; Semb, H.; Kubista, M. Properties of the Reverse Transcription Reaction in mRNA Quantification. Clin. Chem. 2004, 50, 509–515. [Google Scholar] [CrossRef] [Green Version]
- Gerard, G.F.; D’Alessio, J.M. Reverse transcriptase (ec 2.7.7.49). In Enzymes of Molecular Biology; Burrell, M.M., Ed.; Humana Press: Totowa, NJ, USA, 1993; pp. 73–93. [Google Scholar] [CrossRef]
- Stinson, L.; Keelan, J.; Payne, M.S. Identification and removal of contaminating microbial DNA from PCR reagents: Impact on low-biomass microbiome analyses. Lett. Appl. Microbiol. 2018, 68, 2–8. [Google Scholar] [CrossRef]
- Lindén, M.; Thomsen, C.; Grundevik, P.; Jonasson, E.; Andersson, D.; Runnberg, R.; Dolatabadi, S.; Vannas, C.; Santamarίa, M.L.; Fagman, H.; et al. FET family fusion oncoproteins target the SWI / SNF chromatin remodeling complex. Embo Rep. 2019, 20, e45766. [Google Scholar] [CrossRef]
- Wills, Q.F.; Livak, K.J.; Tipping, A.J.; Enver, T.; Goldson, A.J.; Sexton, D.W.; Holmes, C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013, 31, 748–752. [Google Scholar] [CrossRef]
- Ståhlberg, A.; Kubista, M. The workflow of single-cell expression profiling using quantitative real-time PCR. Expert Rev. Mol. Diagn. 2014, 14, 323–331. [Google Scholar] [CrossRef]
- Dar, R.D.; Razooky, B.S.; Singh, A.; Trimeloni, T.V.; Mccollum, J.M.; Cox, C.D.; Simpson, M.; Weinberger, L.S. Transcriptional burst frequency and burst size are equally modulated across the human genome. PNAS 2012, 109, 17454–17459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S. Minimally disturbed, multicycle, and reproducible synchrony using a eukaryotic “baby machine”. BioEssays 2002, 24, 499–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S. Rethinking synchronization of mammalian cells for cell cycle analysis. Cell. Mol. Life Sci. 2003, 60, 1099–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonasson, E.; Andersson, L.; Dolatabadi, S.; Ghannoum, S.; Åman, P.; Ståhlberg, A. Total mRNA Quantification in Single Cells: Sarcoma Cell Heterogeneity. Cells 2020, 9, 759. https://doi.org/10.3390/cells9030759
Jonasson E, Andersson L, Dolatabadi S, Ghannoum S, Åman P, Ståhlberg A. Total mRNA Quantification in Single Cells: Sarcoma Cell Heterogeneity. Cells. 2020; 9(3):759. https://doi.org/10.3390/cells9030759
Chicago/Turabian StyleJonasson, Emma, Lisa Andersson, Soheila Dolatabadi, Salim Ghannoum, Pierre Åman, and Anders Ståhlberg. 2020. "Total mRNA Quantification in Single Cells: Sarcoma Cell Heterogeneity" Cells 9, no. 3: 759. https://doi.org/10.3390/cells9030759
APA StyleJonasson, E., Andersson, L., Dolatabadi, S., Ghannoum, S., Åman, P., & Ståhlberg, A. (2020). Total mRNA Quantification in Single Cells: Sarcoma Cell Heterogeneity. Cells, 9(3), 759. https://doi.org/10.3390/cells9030759