Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Cell Culture
2.3. Cell Fixation and Permeabilization
2.4. Preparation of Cleavable Fluorescent Streptavidin
2.5. Preparation of Biotin-SS-Ab
2.6. Immunofluorescence with CFS
2.7. Signal Amplification
2.8. Fluorophore and Biotin Cleavage
2.9. Streptavidin Blocking
2.10. Quantification of the Fluorophore Cleavage Efficiency
2.11. Quantification of the Biotin Cleavage Efficiency
2.12. Quantification of the Streptavidin Blocking Efficiency
2.13. Multiplexed Protein Analysis in HeLa Cells
2.14. Conventional Immunofluorescence
2.15. Deparaffinization and Antigen Retrieval of FFPE Tissues
2.16. Protein Staining in FFPE Tissues
2.17. Imaging and Data Analysis
3. Results
3.1. Platform Design
3.2. Design and Synthesis of Cleavable Biotin-Conjugated Antibodies and CFS
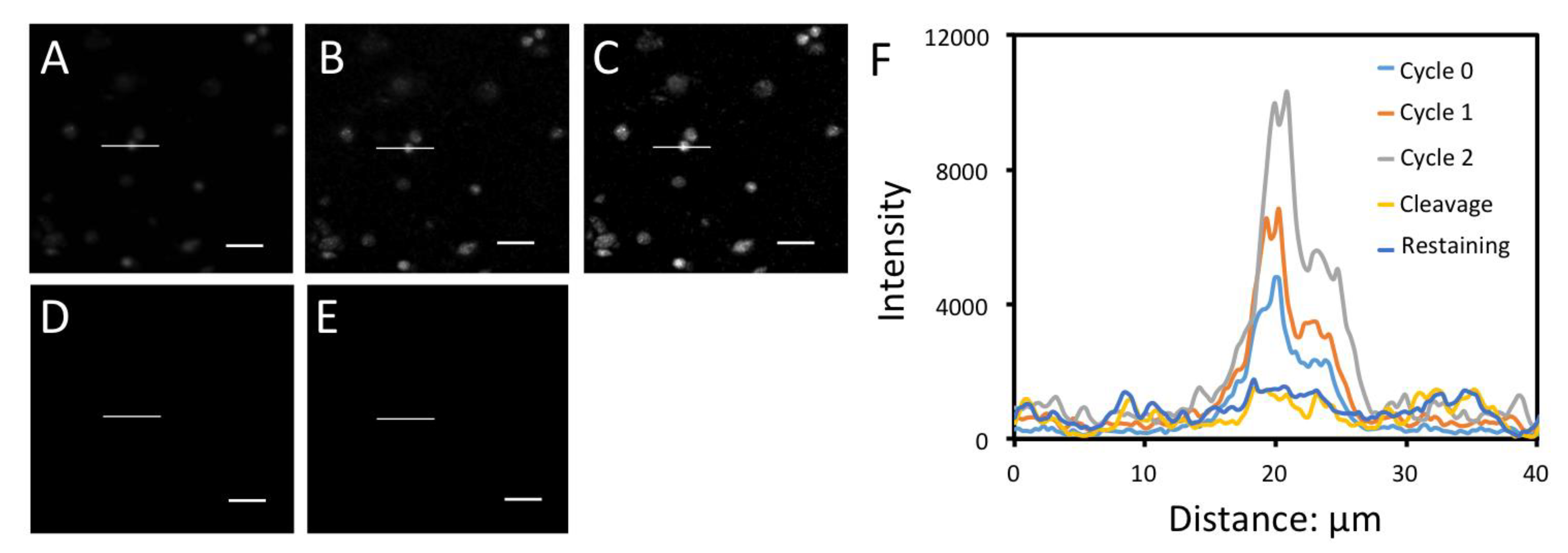
3.3. Significantly Enhanced Detection Sensitivity
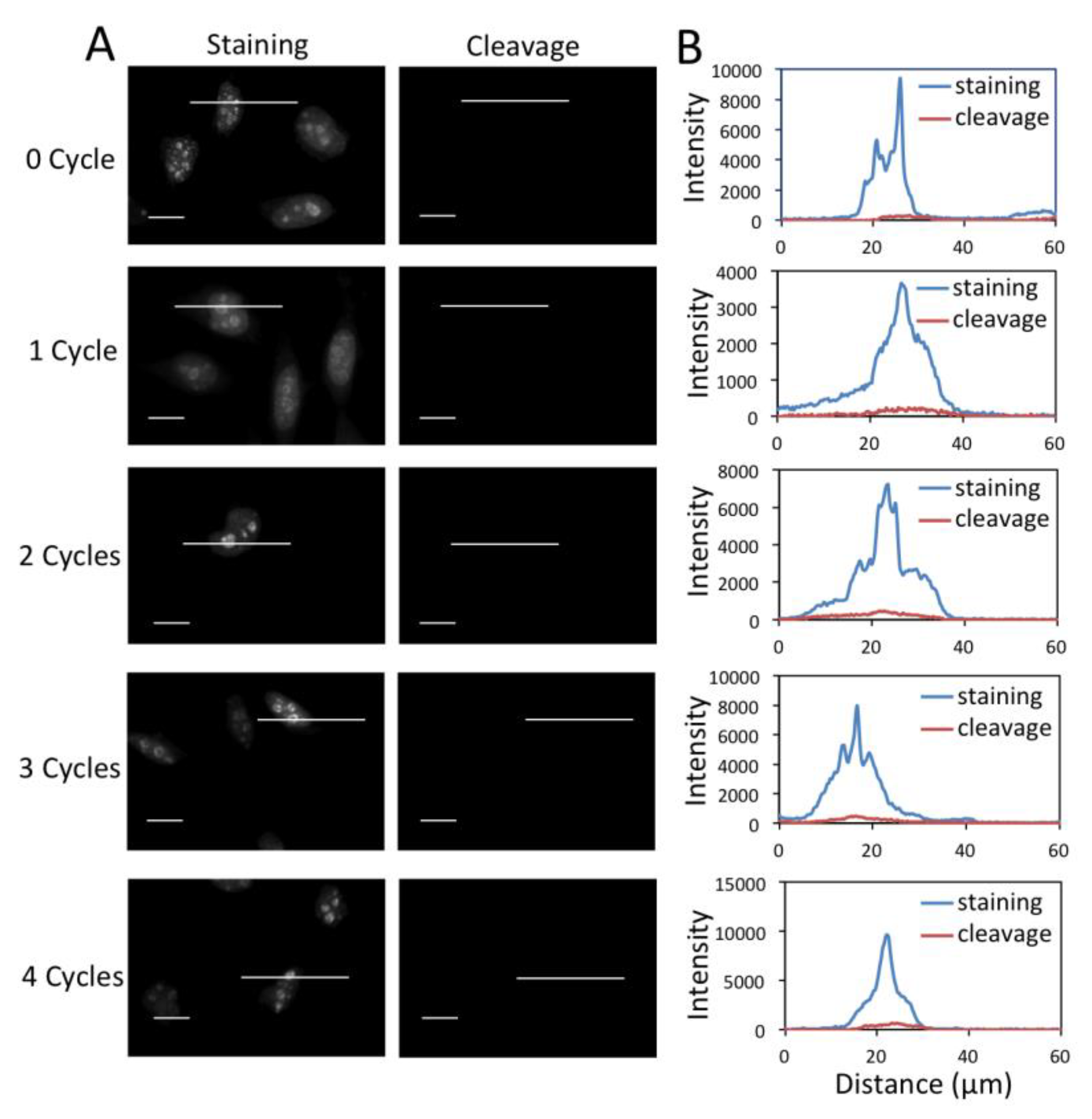
3.4. Efficient Fluorophore and Biotin Cleavage and Streptavidin Blocking
3.5. Multiplexed In-Situ Protein Profiling
3.6. In-Situ Protein Profiling in Human FFPE Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Steininger, R.J.; Rajaram, S.; Girard, L.; Minna, J.D.; Wu, L.F.; Altschuler, S.J. On comparing heterogeneity across biomarkers. Cytom. Part A 2015, 87, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Junker, J.P.; Van Oudenaarden, A. Every cell is special: Genome-wide studies add a new dimension to single-cell biology. Cell 2014, 157, 8–11. [Google Scholar] [CrossRef] [Green Version]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Xue, Q.; Eisele, M.R.; Sulistijo, E.S.; Brower, K.; Han, L.; Amir, E.D.; Pe’er, D.; Miller-Jensen, K.; Fan, R. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc. Natl. Acad. Sci. USA 2015, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, M.J.; Sevinsky, C.J.; Sood, A.; Adak, S.; Bello, M.O. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. USA 2013, 110, 11982–11987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, M.; Liao, R.; Nazaroff, C.D.; Samuel, A.D.; Guo, J. Highly multiplexed single-cell in situ RNA and DNA analysis with bioorthogonal cleavable fluorescent oligonucleotides. Chem. Sci. 2018, 9, 2909–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.Y.; Bindokas, V.P.; Kron, S.J. Multiplex three-dimensional mapping of macromolecular drug distribution in the tumor microenvironment. Mol. Cancer Ther. 2019, 18, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.Y.; Bindokas, V.P.; Kron, S.J. Multiplex three-dimensional optical mapping of tumor immune microenvironment. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Bhowmick, S.; Yu, J.; Wang, J. Highly Multiplexed Single-Cell Protein Profiling with Large-Scale Convertible DNA-Antibody Barcoded Arrays. Adv. Sci. 2018, 1800672, 1800672. [Google Scholar] [CrossRef]
- Shao, S.; Li, Z.; Cheng, H.; Wang, S.; Perkins, N.G.; Sarkar, P.; Wei, W.; Xue, M. A Chemical Approach for Profiling Intracellular AKT Signaling Dynamics from Single Cells. J. Am. Chem. Soc. 2018, 140, 13586–13589. [Google Scholar] [CrossRef]
- Guo, J.; Wang, S.; Dai, N.; Teo, Y.N.; Kool, E.T. Multispectral labeling of antibodies with polyfluorophores on a DNA backbone and application in cellular imaging. Proc. Natl. Acad. Sci. USA 2011, 108, 3493–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, D.; Savage, K.; Reis-Filho, J.S.; Isacke, C.M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded tissue. BMC Mol. Biol. 2008, 9, 1–10. [Google Scholar]
- Mondal, M.; Liao, R.; Xiao, L.; Eno, T.; Guo, J. Highly Multiplexed Single-Cell In Situ Protein Analysis with Cleavable Fluorescent Antibodies. Angew. Chemie Int. Ed. 2017, 56, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Schweller, R.M.; Zimak, J.; Duose, D.Y.; Qutub, A.A.; Hittelman, W.N.; Diehl, M.R. Multiplexed in situ immunofluorescence using dynamic DNA complexes. Angew. Chem. Int. Ed. Engl. 2012, 51, 9292–9296. [Google Scholar] [CrossRef]
- Duose, D.Y.; Schweller, R.M.; Zimak, J.; Rogers, A.R.; Hittelman, W.N.; Diehl, M.R. Configuring robust DNA strand displacement reactions for in situ molecular analyses. Nucleic Acids Res. 2012, 40, 3289–3298. [Google Scholar] [CrossRef]
- Zrazhevskiy, P.; Gao, X. Quantum dot imaging platform for single-cell molecular profiling. Nat. Commun. 2013, 4, 1619. [Google Scholar] [CrossRef]
- Lin, J.R.; Fallahi-Sichani, M.; Sorger, P.K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981.e15. [Google Scholar] [CrossRef] [Green Version]
- Giesen, C.; Wang, H.A.O.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Blow, N. Tissues issues. Nature 2007, 448, 959–962. [Google Scholar] [CrossRef]
- Xie, R.; Chung, J.-Y.; Ylaya, K.; Williams, R.L.; Guerrero, N.; Nakatsuka, N.; Badie, C.; Hewitt, S.M. Factors influencing the degradation of archival formalin-fixed paraffin-embedded tissue sections. J. Histochem. Cytochem. 2011, 59, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Guo, J. Multiplexed single-cell in situ RNA analysis by reiterative hybridization. Anal. Methods 2015, 7, 7290–7295. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Guo, J. Single-Cell in Situ RNA Analysis With Switchable Fluorescent Oligonucleotides. Front. Cell Dev. Biol. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Wei, W.; Su, Y.; Kim, J.; Shin, Y.S.; Mai, W.X.; Nathanson, D.A.; Heath, J.R. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J. Am. Chem. Soc. 2015, 137, 4066–4069. [Google Scholar] [CrossRef] [PubMed] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, R.; Pham, T.; Mastroeni, D.; Coleman, P.D.; Labaer, J.; Guo, J. Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin. Cells 2020, 9, 852. https://doi.org/10.3390/cells9040852
Liao R, Pham T, Mastroeni D, Coleman PD, Labaer J, Guo J. Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin. Cells. 2020; 9(4):852. https://doi.org/10.3390/cells9040852
Chicago/Turabian StyleLiao, Renjie, Thai Pham, Diego Mastroeni, Paul D. Coleman, Joshua Labaer, and Jia Guo. 2020. "Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin" Cells 9, no. 4: 852. https://doi.org/10.3390/cells9040852
APA StyleLiao, R., Pham, T., Mastroeni, D., Coleman, P. D., Labaer, J., & Guo, J. (2020). Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin. Cells, 9(4), 852. https://doi.org/10.3390/cells9040852