Deletion of P2X7 Receptor Decreases Basal Glutathione Level by Changing Glutamate-Glutamine Cycle and Neutral Amino Acid Transporters

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Chemicals
2.2. NAC Treatment and Acute Brain Slices
2.3. GSH Assay
2.4. Immunohisto Chemistry
2.5. Western Blot
2.6. Data Analysis
3. Results
3.1. P2X7R Deletion Increases GS and ASCT2 Expression
3.2. P2X7R Deletion Reduces GSH Concentration
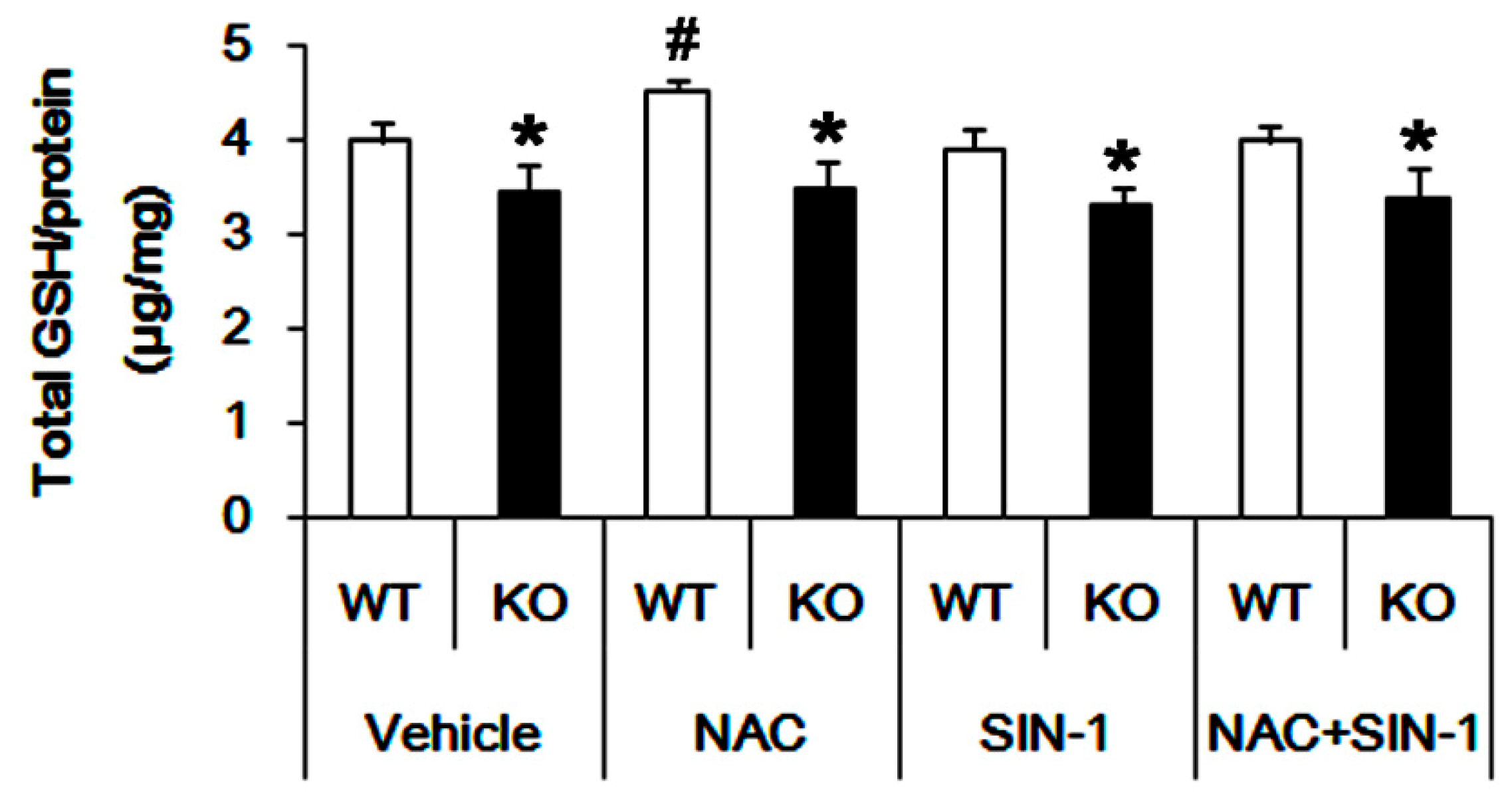
3.3. P2X7R Deletion Inhibits xCT-Mediated NAC Transport
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-dependent detoxification processes in astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Liddell, J.R.; Dringen, R.; Crack, P.J.; Robinson, S.R. Glutathione peroxidase 1 and a high cellular glutathione concentration are essential for effective organic hydroperoxide detoxification in astrocytes. Glia 2006, 54, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000, 62, 649–671. [Google Scholar] [CrossRef]
- Wessner, B.; Strasser, E.M.; Spittler, A.; Roth, E. Effect of single and combined supply of glutamine, glycine, N-acetylcysteine, and R,S-alpha-lipoic acid on glutathione content of myelomonocytic cells. Clin. Nutr. 2003, 22, 515–522. [Google Scholar] [CrossRef]
- Sun, X.; Erb, H.; Murphy, T.H. Coordinate regulation of glutathione metabolism in astrocytes by Nrf2. Biochem. Biophys. Res. Commun. 2005, 326, 371–377. [Google Scholar] [CrossRef]
- Hayashi, M.K. Structure-function relationship of transporters in the glutamate-glutamine cycle of the central nervous system. Int. J. Mol. Sci. 2018, 19, 1177. [Google Scholar] [CrossRef] [Green Version]
- Utsunomiya-Tate, N.; Endou, H.; Kanai, Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 1996, 271, 14883–14890. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Hediger, M.A. The glutamate and neutral amino acid transporter family: Physiological and pharmacological implications. Eur. J. Pharmacol. 2003, 479, 237–247. [Google Scholar] [CrossRef]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef]
- Van Liefferinge, J.; Bentea, E.; Demuyser, T.; Albertini, G.; Follin-Arbelet, V.; Holmseth, S.; Merckx, E.; Sato, H.; Aerts, J.L.; Smolders, I.; et al. Comparative analysis of antibodies to xCT (Slc7a11): Forewarned is forearmed. J. Comp. Neurol. 2016, 524, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kang, T.C. The P2X7 receptor-pannexin-1 complex decreases muscarinic acetylcholine receptor-mediated seizure susceptibility in mice. J. Clin. Invest. 2011, 121, 2037–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor regulates leukocyte infiltrations in rat frontoparietal cortex following status epilepticus. J. Neuroinflamm. 2010, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Codocedo, J.F.; Godoy, J.A.; Poblete, M.I.; Inestrosa, N.C.; Huidobro-Toro, J.P. ATP induces NO production in hippocampal neurons by P2X(7) receptor activation independent of glutamate signaling. PLoS ONE 2013, 8, e57626. [Google Scholar] [CrossRef] [Green Version]
- Ficker, C.; Rozmer, K.; Kató, E.; Andó, R.D.; Schumann, L.; Krügel, U.; Franke, H.; Sperlágh, B.; Riedel, T.; Illes, P. Astrocyte-neuron interaction in the substantia gelatinosa of the spinal cord dorsal horn via P2X7 receptor-mediated release of glutamate and reactive oxygen species. Glia 2014, 62, 1671–1686. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.C.; Huang, W.C.; Chou, Y.C.; Tseng, C.H.; Lee, W.L.; Sun, S.H. Activation of P2X(7) receptors decreases glutamate uptake and glutamine synthetase activity in RBA-2 astrocytes via distinct mechanisms. J. Neurochem. 2008, 105, 151–164. [Google Scholar] [CrossRef]
- Fu, W.; Ruangkittisakul, A.; MacTavish, D.; Baker, G.B.; Ballanyi, K.; Jhamandas, J.H. Activity and metabolism-related Ca2+ and mitochondrial dynamics in co-cultured human fetal cortical neurons and astrocytes. Neuroscience 2013, 250, 520–535. [Google Scholar] [CrossRef]
- Rana, S.; Dringen, R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007, 415, 45–48. [Google Scholar] [CrossRef]
- Stridh, M.H.; Correa, F.; Nodin, C.; Weber, S.G.; Blomstrand, F.; Nilsson, M.; Sandberg, M. Enhanced glutathione efflux from astrocytes in culture by low extracellular Ca2+ and curcumin. Neurochem. Res. 2010, 35, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.C.; Chou, Y.C.; Sun, S.H. P2X7 R-mediated Ca(2+) -independent d-serine release via pannexin-1 of the P2X7 R-pannexin-1 complex in astrocytes. Glia 2015, 63, 877–893. [Google Scholar] [CrossRef] [PubMed]
- Reyes, R.C.; Cittolin-Santos, G.F.; Kim, J.E.; Won, S.J.; Brennan-Minnella, A.M.; Katz, M.; Glass, G.A.; Swanson, R.A. Neuronal glutathione content and antioxidant capacity can be normalized in Situ by N-acetyl cysteine concentrations attained in human cerebrospinal fluid. Neurotherapeutics 2016, 13, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.A.; Cerniglia, G.J.; Zaman, A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal. Biochem. 1990, 190, 360–365. [Google Scholar] [CrossRef]
- Won, S.J.; Kim, J.E.; Cittolin-Santos, G.F.; Swanson, R.A. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J. Neurosci. 2015, 35, 7143–7152. [Google Scholar] [CrossRef] [Green Version]
- Gegelashvili, M.; Rodriguez-Kern, A.; Pirozhkova, I.; Zhang, J.; Sung, L.; Gegelashvili, G. High-affinity glutamate transporter GLAST/EAAT1 regulates cell surface expression of glutamine/neutral amino acid transporter ASCT2 in human fetal astrocytes. Neurochem. Int. 2006, 48, 611–615. [Google Scholar] [CrossRef]
- Bröer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Bröer, S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 1999, 73, 2184–2194. [Google Scholar]
- Tetsuka, K.; Takanaga, H.; Ohtsuki, S.; Hosoya, K.; Terasaki, T. The l-isomer-selective transport of aspartic acid is mediated by ASCT2 at the blood-brain barrier. J. Neurochem. 2003, 87, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Gliddon, C.M.; Shao, Z.; LeMaistre, J.L.; Anderson, C.M. Cellular distribution of the neutral amino acid transporter subtype ASCT2 in mouse brain. J. Neurochem. 2009, 108, 372–383. [Google Scholar] [CrossRef]
- Console, L.; Scalise, M.; Tarmakova, Z.; Coe, I.R.; Indiveri, C. N-linked glycosylation of human SLC1A5 (ASCT2) transporter is critical for trafficking to membrane. Biochim. Biophys. Acta. 2015, 1853, 1636–1645. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, H.; Wu, F.; Qi, Z.; Li, J.; Xu, B.; Liu, W.; Xu, Z.; Deng, Y. Mn inhibits GSH Synthesis via downregulation of neuronal EAAC1 and astrocytic xCT to cause oxidative damage in the striatum of Mice. Oxid. Med. Cell Longev. 2018, 2018, 4235695. [Google Scholar] [CrossRef] [PubMed]
- Nasca, C.; Bigio, B.; Zelli, D.; de Angelis, P.; Lau, T.; Okamoto, M.; Soya, H.; Ni, J.; Brichta, L.; Greengard, P.; et al. Role of the astroglial glutamate exchanger xCT in ventral hippocampus in resilience to stress. Neuron 2017, 96, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.; Bushman, J.; Munger, J.; Noble, M.; Pröschel, C.; Mayer-Pröschel, M. Mutation of ataxia-telangiectasia mutated is associated with dysfunctional glutathione homeostasis in cerebellar astroglia. Glia 2016, 64, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Li, S.; Marshall, Z.M.; Whorton, A.R. A cystine-cysteine shuttle mediated by xCT facilitates cellular responses to S-nitrosoalbumin. Am. J. Physiol. Cell Physiol. 2008, 294, C1012–C1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, Y.C.; Yang, Y.; Tiwari, N.K.; Patrick, B.; Sharma, A.; Li, J.; Awasthi, S. Regulation of 4-hydroxynonenal-mediated signaling by glutathione S-transferases. Free Radic. Biol. Med. 2004, 37, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, Z.A.; Guerra, A.N.; Hill, L.M.; Gavala, M.L.; Prabhu, U.; Aga, M.; Hall, D.J.; Bertics, P.J. Nucleotide receptor signaling in murine macrophages is linked to reactive oxygen species generation. Free Radic. Biol. Med. 2007, 42, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Monção-Ribeiro, L.C.; Cagido, V.R.; Lima-Murad, G.; Santana, P.T.; Riva, D.R.; Borojevic, R.; Zin, W.A.; Cavalcante, M.C.; Riça, I.; Brando-Lima, A.C.; et al. Lipopolysaccharide-induced lung injury: Role of P2X7 receptor. Respir. Physiol. Neurobiol. 2011, 179, 314–325. [Google Scholar] [CrossRef]
- Monção-Ribeiro, L.C.; Faffe, D.S.; Santana, P.T.; Vieira, F.S.; da Graça, C.L.; Marques-da-Silva, C.; Machado, M.N.; Caruso-Neves, C.; Zin, W.A.; Borojevic, R.; et al. P2X7 receptor modulates inflammatory and functional pulmonary changes induced by silica. PLoS ONE 2014, 9, e110185. [Google Scholar] [CrossRef]
- Tonetti, M.; Sturla, L.; Giovine, M.; Benatti, U.; De Flora, A. Extracellular ATP enhances mRNA levels of nitric oxide synthase and TNF-alpha in lipopolysaccharide-treated RAW 264.7 murine macrophages. Biochem. Biophys. Res. Commun. 1995, 214, 125–130. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Bergamaschi, C.T.; Fernandes, M.J.; Paredes-Gamero, E.J.; Buri, M.V.; Ferreira, A.T.; Araujo, S.R.; Punaro, G.R.; Maciel, F.R.; Nogueira, G.B.; et al. P2X(7) receptor in the kidneys of diabetic rats submitted to aerobic training or to N-acetylcysteine supplementation. PLoS ONE 2014, 9, e97452. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the cystine/glutamate antiporter system Xc− in the brain, kidney, and duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBean, G.J.; Flynn, J. Molecular mechanisms of cystine transport. Biochem. Soc. Trans. 2001, 29, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Schrammel, A.; Pfeiffer, S.; Schmidt, K.; Koesling, D.; Mayer, B. Activation of soluble guanylyl cyclase by the nitrovasodilator 3-morpholinosydnonimine involves formation of S-nitrosoglutathione. Mol. Pharmacol. 1998, 54, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Burdo, J.; Schubert, D.; Maher, P. Glutathione production is regulated via distinct pathways in stressed and non-stressed cortical neurons. Brain Res. 2008, 1189, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokołowska, M.; Włodek, L.; Srebro, Z.; Wróbel, M. The effect of nitrogen oxide level modulation on the content of thiol compounds and anaerobic sulfur metabolism in mice brains. Neurobiology 1999, 7, 461–477. [Google Scholar]
- Kopp, R.; Krautloher, A.; Ramírez-Fernández, A.; Nicke, A. P2X7 Interactions and signaling—Making head or tail of it. Front. Mol. Neurosci. 2019, 12, 183. [Google Scholar] [CrossRef]
- Armstrong, S.; Pereverzev, A.; Dixon, S.J.; Sims, S.M. Activation of P2X7 receptors causes isoform-specific translocation of protein kinase C in osteoclasts. J. Cell Sci. 2009, 122, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.D.; Soltoff, S.P. P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem. J. 2002, 366, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Gendron, F.P.; Neary, J.T.; Theiss, P.M.; Sun, G.Y.; Gonzalez, F.A.; Weisman, G.A. Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am. J. Physiol. Cell Physiol. 2003, 284, C571–C581. [Google Scholar] [CrossRef]
- Hung, A.C.; Chu, Y.J.; Lin, Y.H.; Weng, J.Y.; Chen, H.B.; Au, Y.C.; Sun, S.H. Roles of protein kinase C in regulation of P2X7 receptor-mediated calcium signalling of cultured type-2 astrocyte cell line, RBA-2. Cell Signal. 2005, 17, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Brodie, C.; Bogi, K.; Acs, P.; Lorenzo, P.S.; Baskin, L.; Blumberg, P.M. Protein kinase C δ (PKCδ) inhibits the expression of glutamine synthetase in glial cells via the PKCδ regulatory domain and its tyrosine phosphorylation. J. Biol. Chem. 1998, 273, 30713–30718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Fraguela, M.E.; Blanco, L.; Fernández, C.I.; Lorigados, L.; Serrano, T.; Fernández, J.L. Glutathione depletion: Starting point of brain metabolic stress, neuroinflammation and cognitive impairment in rats. Brain Res. Bull. 2018, 137, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, P.; Navarro, S.; Graña-Montes, R.; Bañó-Polo, M.; Fernández, M.R.; Papaleo, E.; Ventura, S. A single cysteine post-translational oxidation suffices to compromise globular proteins kinetic stability and promote amyloid formation. Redox Biol. 2018, 14, 566–575. [Google Scholar] [CrossRef]
- Amorim, R.P.; Araújo, M.G.L.; Valero, J.; Lopes-Cendes, I.; Pascoal, V.D.B.; Malva, J.O.; da Silva Fernandes, M.J. Silencing of P2X7R by RNA interference in the hippocampus can attenuate morphological and behavioral impact of pilocarpine-induced epilepsy. Purinergic Signal. 2017, 13, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Arslan, G.; Avci, B.; Kocacan, S.E.; Rzayev, E.; Ayyildiz, M.; Agar, E. The interaction between P2X7Rs and T-type calcium ion channels in penicillin-induced epileptiform activity. Neuropharmacology 2019, 149, 1–12. [Google Scholar] [CrossRef]
- Soni, N.; Koushal, P.; Reddy, B.V.; Deshmukh, R.; Kumar, P. Effect of GLT-1 modulator and P2X7 antagonists alone and in combination in the kindling model of epilepsy in rats. Epilepsy Behav. 2015, 48, 4–14. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| 4-HNE | Rabbit | Alpha Diagnostic (# HNE11-S) | 1:1000 (IH) |
| ASCT2 | Rabbit | Alomone labs (#ANT-082) | 1:500 (WB) |
| GCLC | Rabbit | Abcam (#ab190685) | 1:2000 (WB) |
| GFAP | Mouse | Millipore (#MAB3402) | 1:1000 (IH) |
| GLS | Rabbit | Abcam (#ab93434) | 1:1000 (WB) |
| GS | Mouse | Millipore (#MAB302) | 1:1000 (WB) |
| GSS | Rabbit | Abcam (#ab133592) | 1:2000 (WB) |
| MAP2 | Mouse | Millipore (#MAB3418) | 1:100 (IH) |
| xCT | Rabbit | Abcam (#ab175186) | 1:1000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:5000 (WB) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Kim, J.-E. Deletion of P2X7 Receptor Decreases Basal Glutathione Level by Changing Glutamate-Glutamine Cycle and Neutral Amino Acid Transporters. Cells 2020, 9, 995. https://doi.org/10.3390/cells9040995
Park H, Kim J-E. Deletion of P2X7 Receptor Decreases Basal Glutathione Level by Changing Glutamate-Glutamine Cycle and Neutral Amino Acid Transporters. Cells. 2020; 9(4):995. https://doi.org/10.3390/cells9040995
Chicago/Turabian StylePark, Hana, and Ji-Eun Kim. 2020. "Deletion of P2X7 Receptor Decreases Basal Glutathione Level by Changing Glutamate-Glutamine Cycle and Neutral Amino Acid Transporters" Cells 9, no. 4: 995. https://doi.org/10.3390/cells9040995
APA StylePark, H., & Kim, J. -E. (2020). Deletion of P2X7 Receptor Decreases Basal Glutathione Level by Changing Glutamate-Glutamine Cycle and Neutral Amino Acid Transporters. Cells, 9(4), 995. https://doi.org/10.3390/cells9040995