Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level



Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Isolation of CTCs Using the ScreenCell Filtration Technique and Immunostaining
2.3. Co-Immuno Telomere Three-Dimensional Quantitative Fluorescent In Situ Hybridization (3-D-QFISH)
2.4. Imaging and Analysis
2.5. Laser Microdissection and Whole-Exome Amplification
2.6. Whole-Exome Sequencing and Bioinformatics Analysis
3. Results
3.1. High-Risk Prostate Cancer CTCs Were Selected Based on Their Positive Staining for the Androgen Receptor and Cytokeratin 8, 18, and 19 and Negativity for CD45.
3.2. CTCs from Localized High-Risk Prostate Patients Showed Telomere-Related Heterogeneity at the Single-Cell Level
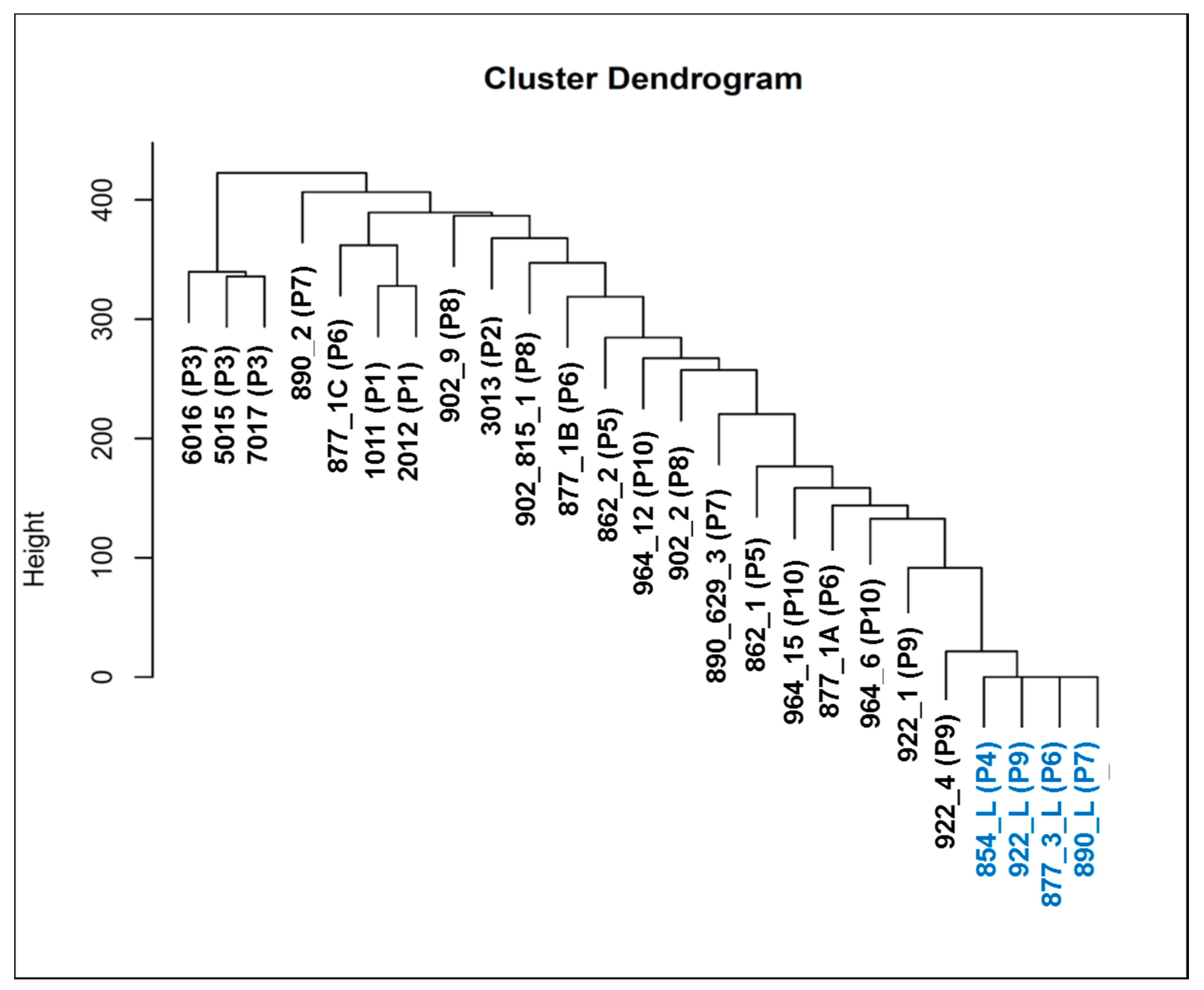
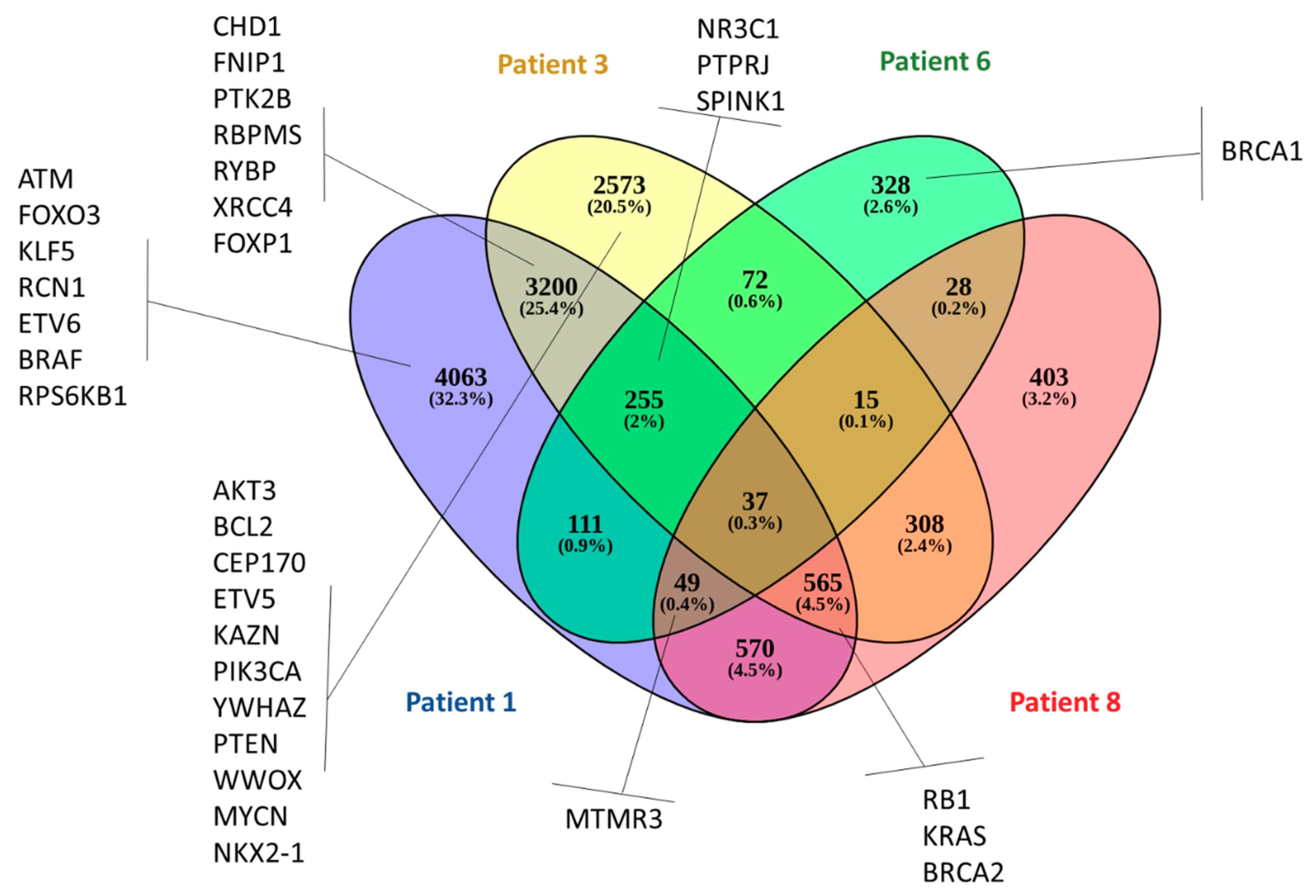
3.3. Whole-Exome Sequencing Showed Genetic Variation (SNVs and Indels) Associated with Telomere Maintenance Genes, Prostate Cancer, and Known Cancer Drug Response
3.4. Copy Number Alterations Identify Gene Amplifications Associated with High-Risk Prostate CTCs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef]
- Kattan, M.W.; Eastham, J.A.; Stapleton, A.M.; Wheeler, T.M.; Scardino, P.T. A preoperative nomogram for disease recurrence following radical prostatectomy for prostate cancer. J. Natl. Cancer Inst. 1998, 90, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU guidelines on prostate cancer. part 1: Screening, diagnosis, and local treatment with curative intent-update 2013. Eur. Urol. 2014, 65, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Choyke, P.L.; Figg, W.D.; Turkbey, B. The role of MRI in prostate cancer active surveillance. Biomed. Res. Int. 2014, 2014, 203906. [Google Scholar] [CrossRef] [PubMed]
- Kgatle, M.M.; Kalla, A.A.; Islam, M.M.; Sathekge, M.; Moorad, R. Prostate Cancer: Epigenetic Alterations, Risk Factors, and Therapy. Prostate Cancer 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.C.; Rumble, R.B.; Loblaw, D.A.; Finelli, A.; Ehdaie, B.; Cooperberg, M.R.; Morgan, S.C.; Tyldesley, S.; Haluschak, J.J.; Tan, W.; et al. Active Surveillance for the Management of Localized Prostate Cancer (Cancer Care Ontario Guideline): American Society of Clinical Oncology Clinical Practice Guideline Endorsement. J. Clin. Oncol. 2016, 34, 2182–9210. [Google Scholar] [CrossRef] [Green Version]
- Wilt, T.J.; Andriole, G.L.; Brawer, M.K. Prostatectomy versus Observation for Early Prostate Cancer. N. Engl. J. Med. 2017, 377, 1302–1303. [Google Scholar] [CrossRef] [PubMed]
- Punnen, S.; Cooperberg, M.R.; D’Amico, A.V.; Karakiewicz, P.I.; Moul, J.W.; Scher, H.I.; Schlomm, T.; Freedland, S.J. Management of biochemical recurrence after primary treatment of prostate cancer: A systematic review of the literature. Eur. Urol. 2013, 64, 905–915. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, J.F.; Sabelnykova, V.Y.; Weischenfeldt, J.; Simon, R.; Aguiar, J.A.; Alkallas, R.; Heisler, L.E.; Zhang, J.; Watson, J.D.; Chua, M.L.K.; et al. Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 2017, 8, 656. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, S.M.G.; Liu, L.Y.; Rubanova, Y.; Bhandari, V.; Holgersen, L.E.; Szyca, L.M.; Fox, N.; Chua, M.L.K.; Yamaguchi, T.N.; Heisler, L.E. The Evolutionary Landscape of Localized Prostate Cancers Drives Clinical Aggression. Cell 2018, 173, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- McCrow, J.P.; Petersen, D.C.; Louw, M.; Chan, E.K.; Harmeyer, K.; Vecchiarelli, S.; Lyons, R.J.; Bornman, M.S.; Hayes, V.M. Spectrum of mitochondrial genomic variation and associated clinical presentation of prostate cancer in South African men. Prostate 2016, 76, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Maki, J.; Robinson, K.; Reguly, B.; Alexander, J.; Wittock, R.; Aguirre, A.; Diamandis, E.P.; Escott, N.; Skehan, A.; Prowse, O.; et al. Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am. J. Clin. Pathol. 2008, 129, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Pantel, K. Liquid biopsies, what we do not know (yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.L.; Christie, M. Circulating tumor DNA analysis detects, minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.L.; Lee, R.J.; Nagrath, S.; Yu, M.; Miyamoto, D.T.; Ulkus, L.; Inserra, E.J.; Ulman, M.; Springer, S.; Nakamura, Z. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2010, 2, 25ra23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.W.; Nakanishi, H.; Kumar, V.S.; Bhadkamkar, V.A.; McCormack, R.; Fritsche, H.A.; Handy, B.; Gornet, T.; Babaian, R.J. Circulating tumor cells in peripheral blood samples from patients with increased serum prostate specific antigen: Initial results in early prostate cancer. J. Urol. 2008, 179, 2187–2191. [Google Scholar] [CrossRef]
- Meyer, C.P.; Pantel, K.; Tennstedt, P.; Stroelin, P.; Schlomm, T.; Heinzer, H.; Riethdorf, S.; Steuber, T. Limited prognostic value of preoperative circulating tumor cells for early biochemical recurrence in patients with localized prostate cancer. In Urologic Oncology: Seminars and Original Investigations; Elviser: Amsterdam, The Netherlands, 2016; Volume 34, pp. 235.e11–235.e16. [Google Scholar] [CrossRef]
- Liu, W.; Yin, B.; Wang, X.; Yu, P.; Duan, X.; Liu, C.; Wang, B.; Tao, Z. Circulating tumor cells in prostate cancer: Precision diagnosis and therapy. Oncol. Lett. 2017, 14, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- DeSitter, I.; Guerrouahen, B.S.; Benali-Furet, N.; Wechsler, J.; Jänne, P.A.; Kuang, Y.; Yanagita, M.; Wang, L.; Berkowitz, J.A.; Distel, R.J.; et al. A new device for rapid isolation by size and characterization of rare circulating tumor cells. Anticancer Res. 2011, 31, 427–441. [Google Scholar]
- Drachenberg, D.; Awe, J.A.; Rangel-Pozzo, A.; Saranchuk, J.; Mai, S. Advancing Risk Assessment of Intermediate Risk Prostate Cancer Patients. Cancers 2019, 11, 855. [Google Scholar] [CrossRef] [Green Version]
- Wark, L.; Quon, H.; Ong, A.; Drachenberg, D.; Rangel-Pozzo, A.; Mai, S. Long-Term Dynamics of Three Dimensional Telomere Profiles in Circulating Tumor Cells in High-Risk Prostate Cancer Patients Undergoing Androgen-Deprivation and Radiation Therapy. Cancers 2019, 11, 1165. [Google Scholar] [CrossRef] [Green Version]
- Graham, M.K.; Meeker, A. Telomeres and telomerase in prostate cancer development and therapy. Nat. Rev. Urol. 2017, 14, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Adebayo, A.J.; Xu, M.C.; Wechsler, J.; Benali-Furet, N.; Cayre, Y.E.; Saranchuk, J.; Drachenberg, D.; Mai, S. Three-Dimensional Telomeric Analysis of Isolated Circulating Tumor Cells (CTCs) Defines CTC Subpopulations. Transl. Oncol. 2013, 6, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Contu, F.; Rangel-Pozzo, A.; Trokajlo, P.; Wark, L.; Klewes, L.; Johnson, N.A.; Petrogiannis-Haliotis, T.; Gartner, J.G.; Garini, Y.; Vanni, R.; et al. Distinct 3D Structural Patterns of Lamin A/C Expression in Hodgkin and Reed-Sternberg Cells. Cancers 2018, 10, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel-Pozzo, A.; Corrêa de Souza, D.; Schmid-Braz, A.T.; de Azambuja, A.P.; Ferraz-Aguiar, T.; Borgonovo, T.; Mai, S. 3D Telomere Structure Analysis to Detect Genomic Instability and Cytogenetic Evolution in Myelodysplastic Syndromes. Cells 2019, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awe, J.A.; Saranchuk, J.; Drachenberg, D.; Mai, S. Filtration-based enrichment of circulating tumor cells from all prostate cancer risk groups. Urol. Oncol. 2017, 35, 300–309. [Google Scholar] [CrossRef]
- Vermolen, B.J.; Garini, Y.; Mai, S.; Mougey, V.; Fest, T.; Chuang, T.C.; Chuang, A.Y.; Wark, L.; Young, I.T. Characterizing the three-dimensional organization of telomeres. Cytom. A 2005, 67, 144–150. [Google Scholar] [CrossRef]
- Krueger, F. Trim Galore. 2015. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 29 April 2019).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute. Picard Tools. 2018. Available online: http://broadinstitute.github.io/picard (accessed on 16 March 2019).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A Tool for Assessing Copy Number and Allelic Content Using Next-Generation Sequencing Data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Haw, R. Functional Interaction Network Construction and Analysis for Disease Discovery. Methods Mol. Biol. 2017, 1558, 235–253. [Google Scholar] [CrossRef]
- Whirl-Carrillo, M.; McDonagh, E.M.; Hebert, J.M.; Gong, L.; Sangkuhl, K.; Thorn, C.F.; Altman, R.B.; Klein, T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharm. 2012, 92, 414–417. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Ni, X.; Guo, H.; Su, Z.; Ba, Y.; Tong, Z.; Guo, Z.; Yao, X.; Chen, X.; Yin, J.; et al. Single-cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Res. 2017, 27, 1312–1322. [Google Scholar] [CrossRef]
- Friedlander, T.W.; Welty, C.; Anantharaman, A.; Schonhoft, J.D.; Jendrisak, A.; Lee, J.; Li, P.; Hough, J.; Stromlund, A.; Edwards, M. Identification and Characterization of Circulating Tumor Cells in Men Who have Undergone Prostatectomy for Clinically Localized, High Risk Prostate Cancer. J. Urol. 2019, 202, 732–741. [Google Scholar] [CrossRef]
- Ikeda, S.; Elkin, S.K.; Tomson, B.N.; Carter, J.L.; Kurzrock, R. Next-generation Sequencing of Prostate Cancer: Genomic and Pathway Alterations, Potential Actionability Patterns, and Relative Rate of Use of Clinical-Grade Testing. Cancer Biol. 2019, 20, 219–226. [Google Scholar] [CrossRef]
- Wu, Z.; Li, S.; Tang, X.; Wang, Y.; Guo, W.; Cao, G.; Chen, K.; Zhang, M.; Guan, M.; Yang, D. Copy Number Amplification of DNA Damage Repair Pathways Potentiates Therapeutic Resistance in Cancer. Theranostics 2020, 10, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Ma, X.; Zhang, Q.; Wu, C.J.; Liao, W.; Li, J.; Wang, H.; Zhao, J.; Zhou, X.; Guan, C.; et al. USP21 deubiquitinase promotes pancreas cancer cell stemness via Wnt pathway activation. Genes Dev. 2019, 33, 1361–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.C.; Levy, S.; Huang, J.; Stockwell, T.B.; Walenz, B.P.; Li, K.; Axelrod, N.; Busam, D.A.; Strausberg, R.; Venter, J.C. Genetic variation in an individual human exome. PLoS Genet. 2008, 4, e1000160. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Xiong, W.; Li, J.; Wang, X.; Xu, C.; Yang, L. Intersectin 1 (ITSN1) identified by comprehensive bioinformatic analysis and experimental validation as a key candidate biological target in breast cancer. OncoTargets Ther. 2019, 12, 7079–7093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, H.; Kuboshima, M.; Shiratori, T.; Nabeya, Y.; Takeuchi, A.; Takagi, H.; Nomura, F.; Takiguchi, M.; Ochiai, T.; Hiwasa, T. Serum anti-myomegalin antibodies in patients with esophageal squamous cell carcinoma. Int. J. Oncol. 2007, 30, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strogolova, V.; Furness, A.; Robb-McGrath, M.; Garlich, J.; Stuart, R. Rcf1 and Rcf2, Members of the Hypoxia-Induced Gene 1 Protein Family, Are Critical Components of the Mitochondrial Cytochrome bc1-Cytochrome c Oxidase Supercomplex. Mol. Cell. Biol. 2012, 32, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Smogorzewska, A.; De Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, S.A.; Hu, C.; Sartor, O.; Gomella, L.G.; Amin, M.B.; Purdy, J.; Michalski, J.M.; Garzotto, M.G.; Pervez, N.; Balogh, A.G.; et al. Effect of Chemotherapy With Docetaxel With Androgen Suppression and Radiotherapy for Localized High-Risk Prostate Cancer: The Randomized Phase III NRG Oncology RTOG 0521 Trial. J. Clin. Oncol. 2019, 37, 1159–1168. [Google Scholar] [CrossRef]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Xiao, H.; Yang, Q.; Hu, R.; Jiang, L.; Bi, R.; Jiang, X.; Wang, L.; Mei, J.; Ding, F.; et al. The USP21/YY1/SNHG16 axis contributes to tumor proliferation, migration, and invasion of non-small-cell lung cancer. Exp. Mol. Med. 2020, 52, 41–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligthart, S.T.; Coumans, F.A.W.; Bidard, F.; Simkens, L.H.J.; Punt, C.J.A.; Groot, M.R.; Attard, G.; de Bono, J.S.; Pierga, J.; Terstappen, L.W.M.M. Circulating Tumor Cells Count and Morphological Features in Breast, Colorectal and Prostate Cancer. PLoS ONE 2013, 8, e67148. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Patient Number | Patient ID | Type |
|---|---|---|---|
| 1011 | 1 | 806 | CTC |
| 2012 | 1 | 806 | CTC |
| 3013 | 2 | 810 | CTC |
| 5015 | 3 | 823 | CTC |
| 6016 | 3 | 823 | CTC |
| 7017 | 3 | 823 | CTC |
| 862_1 | 5 | 862 | CTC |
| 862_2 | 5 | 862 | CTC |
| 877_1A | 6 | 877 | CTC |
| 877_1B | 6 | 877 | CTC |
| 877_1C | 6 | 877 | CTC |
| 890_629_3 | 7 | 890 | CTC |
| 890_2 | 7 | 890 | CTC |
| 902_815_1 | 8 | 902 | CTC |
| 902_2 | 8 | 902 | CTC |
| 902_9 | 8 | 902 | CTC |
| 922_1 | 9 | 922 | CTC |
| 922_4 | 9 | 922 | CTC |
| 964_12 | 10 | 964 | CTC |
| 964_15 | 10 | 964 | CTC |
| 964_6 | 10 | 964 | CTC |
| 922_L | 9 | 922 | Lymphocyte |
| 877_3_L | 6 | 877 | Lymphocyte |
| 890_L | 7 | 890 | Lymphocyte |
| 854_L | 4 | 854 | Lymphocyte |
| Unique Total | Non-Coding Regions | Coding Regions | |
|---|---|---|---|
| SNVs | 202,241 | 192,129 (95%) | 10,112 (5%) |
| Indels | 137,407 | 127,789 (93%) | 9618 (7%) |
| SNVs | INDELs | |||||
|---|---|---|---|---|---|---|
| CTC Sample | Total | Unannotated | Annotated | Total | Unannotated | Annotated |
| 1011 (P1) | 82,697 | 1034 | 81,663 | 35,204 | 773 | 34,431 |
| 2012 (P1) | 82,469 | 952 | 81,517 | 36,558 | 744 | 35,814 |
| 3013 (P2) | 96,223 | 1118 | 95,105 | 41,631 | 971 | 40,660 |
| 5015 (P3) | 43,373 | 1254 | 42,119 | 32,658 | 13,175 | 19,483 |
| 6016 (P3) | 47,747 | 1054 | 46,693 | 38,159 | 14,555 | 23,604 |
| 7017 (P3) | 46,047 | 1139 | 44,908 | 37,729 | 15,468 | 22,261 |
| 862_1 (P5) | 44,785 | 653 | 44,132 | 32,845 | 4309 | 28,536 |
| 862_2 (P5) | 51,085 | 1222 | 49,863 | 45,574 | 16,026 | 29,548 |
| 877_1A (P6) | 81,783 | 1420 | 80,363 | 49,401 | 12,387 | 37,014 |
| 877_1B (P6) | 5052 | 193 | 4859 | 4261 | 1607 | 2654 |
| 877_1C (P6) | 16,818 | 229 | 16,589 | 12,077 | 1775 | 10,302 |
| 890_629_3 (P7) | 21,506 | 655 | 20,851 | 19,648 | 6462 | 13,186 |
| 890_2 (P7) | 56,421 | 1370 | 55,051 | 43,329 | 16,340 | 26,989 |
| 902_815_1 (P8) | 36,729 | 532 | 36,197 | 19,511 | 2108 | 17,403 |
| 902_2 (P8) | 8380 | 338 | 8042 | 7789 | 2988 | 4801 |
| 902_9 (P8) | 27,091 | 1042 | 26,049 | 27,608 | 11,530 | 16,078 |
| 922_1 (P9) | 4503 | 276 | 4227 | 9596 | 3913 | 5683 |
| 922_4 (P9) | 386 | 154 | 232 | 596 | 525 | 71 |
| 964_12 (P10) | 5441 | 332 | 5109 | 4790 | 2229 | 2561 |
| 964_15 (P10) | 16,671 | 539 | 16,132 | 13,049 | 4649 | 8400 |
| 964_6 (P10) | 7500 | 393 | 7107 | 10,771 | 3844 | 6927 |
| Term | p-Value | Genes |
|---|---|---|
| Generic Transcription Pathway Homo sapiens R-HSA-212436 | 6.28 × 10−4 | ZFP14; ZNF461; PARP1; ZNF382; ZNF529; ZNF566; TEAD1 |
| POLB-Dependent Long Patch Base Excision Repair Homo sapiens R-HSA-110362 | 0.01288 | PARP1 |
| Regulation of cytoskeletal remodeling and cell spreading by IPP complex components Homo sapiens R-HSA-446388 | 0.014707 | PARVA |
| HDR through MMEJ (alt-NHEJ) Homo sapiens R-HSA-5685939 | 0.018351 | PARP1 |
| Dectin-2 family Homo sapiens R-HSA-5621480 | 0.018351 | FCER1G |
| PPARA activates gene expression Homo sapiens R-HSA-1989781 | 0.018526 | APOA2;TEAD1 |
| Regulation of lipid metabolism by Peroxisome proliferator-activated receptor alpha (PPARalpha) Homo sapiens R-HSA-400206 | 0.01946 | APOA2;TEAD1 |
| Heme biosynthesis Homo sapiens R-HSA-189451 | 0.020168 | PPOX |
| Serotonin receptors Homo sapiens R-HSA-390666 | 0.021981 | HTR4 |
| Platelet Adhesion to exposed collagen Homo sapiens R-HSA-75892 | 0.023792 | FCER1G |
| TNFR1-induced proapoptotic signaling Homo sapiens R-HSA-5357786 | 0.023792 | USP21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangel-Pozzo, A.; Liu, S.; Wajnberg, G.; Wang, X.; Ouellette, R.J.; Hicks, G.G.; Drachenberg, D.; Mai, S. Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level. Cells 2020, 9, 1863. https://doi.org/10.3390/cells9081863
Rangel-Pozzo A, Liu S, Wajnberg G, Wang X, Ouellette RJ, Hicks GG, Drachenberg D, Mai S. Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level. Cells. 2020; 9(8):1863. https://doi.org/10.3390/cells9081863
Chicago/Turabian StyleRangel-Pozzo, Aline, Songyan Liu, Gabriel Wajnberg, Xuemei Wang, Rodney J. Ouellette, Geoffrey G. Hicks, Darrel Drachenberg, and Sabine Mai. 2020. "Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level" Cells 9, no. 8: 1863. https://doi.org/10.3390/cells9081863
APA StyleRangel-Pozzo, A., Liu, S., Wajnberg, G., Wang, X., Ouellette, R. J., Hicks, G. G., Drachenberg, D., & Mai, S. (2020). Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level. Cells, 9(8), 1863. https://doi.org/10.3390/cells9081863