Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism



{kind=link}
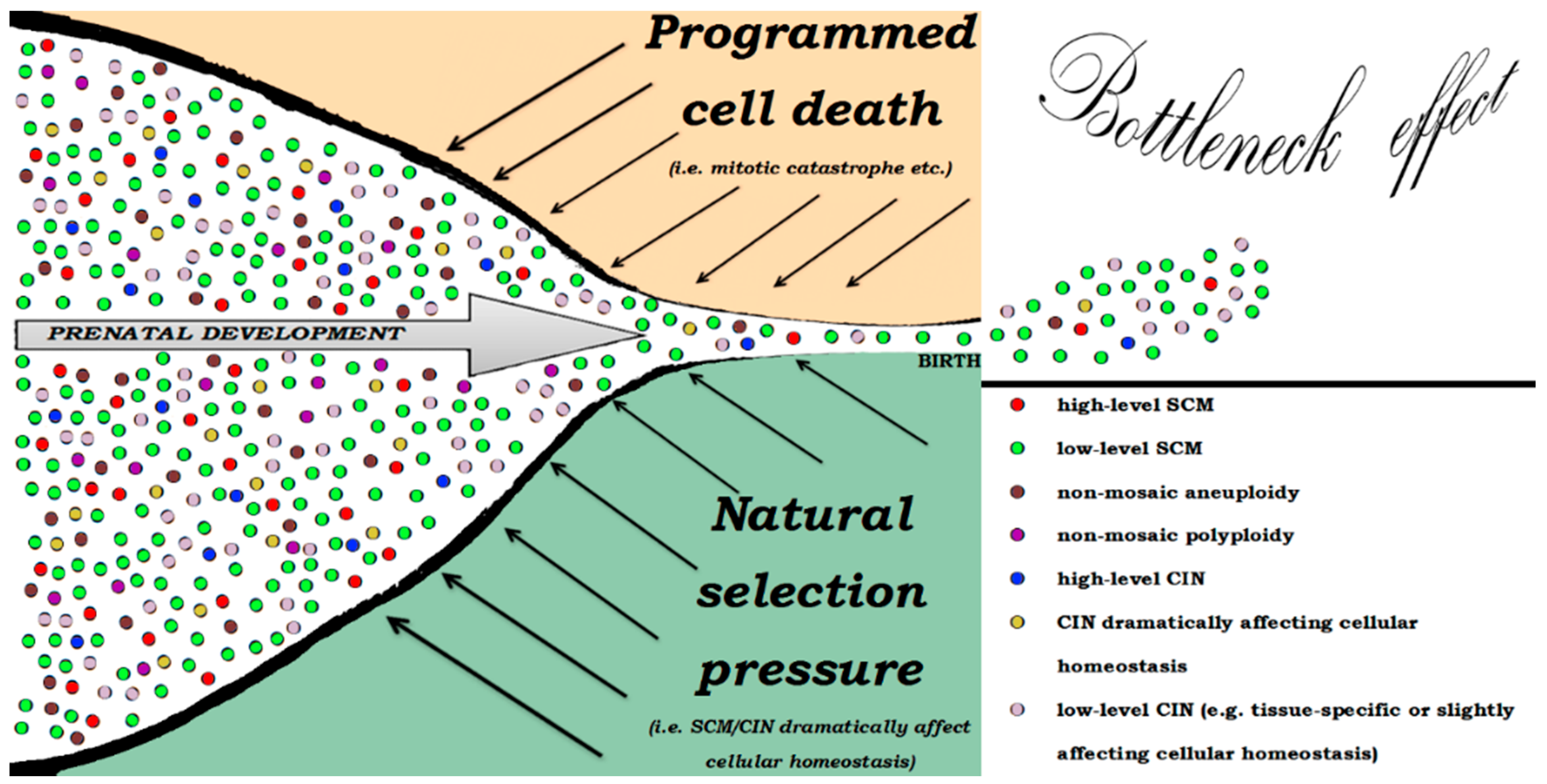{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Chromosomal Mosaicism during Early Development
3. Tissue-Specific Chromosomal Mosaicism
4. Mosaic Chromosome Abnormalities
5. Chromosomal Heterogeneity, Somatic Mosaicism and Human Disease
6. Technical Aspects of SCM/CIN Studies
7. Chromosomal Mosaicism and Aging
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trask, B.J. Human cytogenetics: 46 chromosomes, 46 years and counting. Nat. Rev. Genet. 2002, 3, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, M.A. History and evolution of cytogenetics. Mol. Cytogenet. 2015, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Somatic genome variations in health and disease. Curr. Genom. 2010, 11, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G. Review and hypotheses, somatic mosaicism, observations related to clinical genetics. Am. J. Hum. Genet. 1988, 43, 355–363. [Google Scholar]
- Youssoufian, H.; Pyeritz, R.E. Mechanisms and consequences of somatic mosaicism in humans. Nat. Rev. Genet. 2002, 3, 748–758. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal variation in mammalian neuronal cells, known facts and attractive hypotheses. Int. Rev. Cytol. 2006, 249, 143–191. [Google Scholar]
- Campbell, I.M.; Shaw, C.A.; Stankiewicz, P.; Lupski, J.R. Somatic mosaicism, implications for disease and transmission genetics. Trends Genet. 2015, 31, 382–392. [Google Scholar] [CrossRef]
- Heng, H.H. Missing heritability and stochastic genome alterations. Nat. Rev. Genet. 2010, 11, 813. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J. Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 2014, 26, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.J.; Regan, S.; Liu, G.; Alemara, S.; Heng, H.H. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol. Cytogenet. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1, 26. [Google Scholar] [CrossRef]
- Taylor, T.H.; Gitlin, S.A.; Patrick, J.L.; Crain, J.L.; Wilson, J.M.; Griffin, D.K. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum. Reprod. Update 2014, 20, 571–581. [Google Scholar] [CrossRef] [Green Version]
- Santaguida, S.; Amon, A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef]
- Chunduri, N.K.; Storchová, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Ontogenetic variation of the human genome. Curr. Genom. 2010, 11, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, S.I.; Hassold, T.J.; Hunt, P.A. Human aneuploidy: Mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 2012, 13, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Vanneste, E.; Voet, T.; Le Caignec, C.; Ampe, M.; Konings, P.; Melotte, C.; Debrock, S.; Amyere, M.; Vikkula, M.; Schuit, F.; et al. Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 2009, 15, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, C.; Vanneste, E.; Pexsters, A.; D’Hooghe, T.; Voet, T.; Vermeesch, J.R. Somatic genomic variations in early human prenatal development. Curr. Genom. 2010, 11, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Diez, C.; FitzHarris, G. Causes and consequences of chromosome segregation error in preimplantation embryos. Reproduction 2018, 155, R63–R76. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Yuan, B.; Carvalho, C.M.B.; Wuster, A.; Walter, K.; Zhang, L.; Gambin, T.; Chong, Z.; Campbell, I.M.; Coban Akdemir, Z.; et al. An organismal CNV mutator phenotype restricted to early human development. Cell 2017, 168, 830–842.e7. [Google Scholar] [CrossRef]
- Babariya, D.; Fragouli, E.; Alfarawati, S.; Spath, K.; Wells, D. The incidence and origin of segmental aneuploidy in human oocytes and preimplantation embryos. Hum. Reprod. 2017, 32, 2549–2560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Vijg, J. Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 2018, 52, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Iourov, I.Y.; Monakhov, V.V.; Soloviev, I.V.; Vostrikov, V.M.; Vorsanova, S.G. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J. Histochem. Cytochem. 2005, 53, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Kutsev, S.I.; Pellestor, F.; Beresheva, A.K.; Demidova, I.A.; Kravets, V.S.; et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2007, 2, e558. [Google Scholar] [CrossRef] [PubMed]
- Kalousek, D.K.; Vekemans, M. Confined placental mosaicism. J. Med. Genet. 1996, 33, 529–533. [Google Scholar] [CrossRef]
- Toutain, J.; Goutte-Gattat, D.; Horovitz, J.; Saura, R. Confined placental mosaicism revisited, Impact on pregnancy characteristics and outcome. PLoS ONE 2018, 13, e0195905. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Kolotii, A.D.; Iourov, I.Y.; Monakhov, V.V.; Kirillova, E.A.; Soloviev, I.V.; Yurov, Y.B. Evidence for high frequency of chromosomal mosaicism in spontaneous abortions revealed by interphase FISH analysis. J. Histochem. Cytochem. 2005, 53, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.M.; van Maarle, M.C.; van Wely, M.; Goddijn, M. Genetics of early miscarriage. Biochim. Biophys. Acta 2012, 1822, 1951–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, R.; Sessa, A.M.; Fumo, R.; Gaeta, S. Chromosomal anomalies in early spontaneous abortions: Interphase FISH analysis on 855 FFPE first trimester abortions. Prenat. Diagn. 2016, 36, 186–191. [Google Scholar] [CrossRef]
- Vera-Rodriguez, M.; Chavez, S.L.; Rubio, C.; Reijo Pera, R.A.; Simon, C. Prediction model for aneuploidy in early human embryo development revealed by single-cell analysis. Nat. Commun. 2015, 6, 7601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, R.C. Mosaicism in preimplantation human embryos: When chromosomal abnormalities are the norm. Trends Genet. 2017, 33, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.Y.; Perlis, T.E. United States survey on chromosome mosaicism and pseudomosaicism in prenatal diagnosis. Prenat. Diagn. 1984, 4, 97–130. [Google Scholar] [CrossRef] [PubMed]
- Grati, F.R. Chromosomal mosaicism in human feto-placental development: Implications for prenatal diagnosis. J. Clin. Med. 2014, 3, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Hultén, M.A.; Jonasson, J.; Iwarsson, E.; Uppal, P.; Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Trisomy 21 mosaicism: We may all have a touch of Down syndrome. Cytogenet. Genome Res. 2013, 139, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Horne, S.D.; Chowdhury, S.K.; Heng, H.H. Stress, genomic adaptation, and the evolutionary trade-off. Front. Genet. 2014, 5, 92. [Google Scholar] [CrossRef]
- Nielsen, J.; Wohlert, M. Chromosome abnormalities found among 34,910 newborn children: Results from a 13-year incidence study in Arhus, Denmark. Hum. Genet. 1991, 87, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Qiu, L.Q.; Ye, Y.H.; Xu, J. Chromosomal abnormalities: Subgroup analysis by maternal age and perinatal features in Zhejiang province of China, 2011–2015. Ital. J. Pediatr. 2017, 43, 47. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Molecular cytogenetics and cytogenomics of brain diseases. Curr. Genom. 2008, 9, 452–465. [Google Scholar] [CrossRef]
- Schinzel, A. Catalogue of Unbalanced Chromosome Aberrations in Man; Walter de Gruyter: Berlin, Germany, 2001. [Google Scholar]
- Smith, C.L.; Bolton, A.; Nguyen, G. Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr. Genom. 2010, 11, 447–469. [Google Scholar] [CrossRef]
- Hochstenbach, R.; Buizer-Voskamp, J.E.; Vorstman, J.A.S.; Ophoff, R.A. Genome arrays for the detection of copy number variations in idiopathic mental retardation, idiopathic generalized epilepsy and neuropsychiatric disorders: Lessons for diagnostic workflow and research. Cytogenet. Genome Res. 2011, 135, 174–202. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Yurov, Y.B.; Soloviev, I.V.; Iourov, I.Y. Molecular cytogenetic diagnosis and somatic genome variations. Curr. Genom. 2010, 11, 440–446. [Google Scholar] [CrossRef]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- D’Gama, A.M.; Walsh, C.A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 2018, 21, 1504–1514. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y.; Demidova, I.A.; Beresheva, A.K.; Kravetz, V.S.; Monakhov, V.V.; Kolotii, A.D.; Voinova-Ulas, V.Y.; Gorbachevskaya, N.L. Unexplained autism is frequently associated with low-level mosaic aneuploidy. J. Med. Genet. 2007, 44, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Jauniaux, E.; Burton, G.J. Pathophysiology of histological changes in early pregnancy loss. Placenta 2005, 26, 114–123. [Google Scholar] [CrossRef]
- Mantikou, E.; Wong, K.M.; Repping, S.; Mastenbroek, S. Molecular origin of mitotic aneuploidies in preimplantation embryos. Biochim. Biophys. Acta 2012, 1822, 1921–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varetti, G.; Pellman, D.; Gordon, D.J. Aurea mediocritas: The importance of a balanced genome. Cold Spring Harb. Perspect. Biol. 2014, 6, a015842. [Google Scholar] [CrossRef]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef]
- Weier, J.F.; Weier, H.U.; Jung, C.J.; Gormley, M.; Zhou, Y.; Chu, L.W.; Genbacev, O.; Wright, A.A.; Fisher, S.J. Human cytotrophoblasts acquire aneuploidies as they differentiate to an invasive phenotype. Dev. Biol. 2005, 279, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Intercellular genomic (chromosomal) variations resulting in somatic mosaicism: Mechanisms and consequences. Curr. Genom. 2006, 7, 435–446. [Google Scholar] [CrossRef]
- De, S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011, 27, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, C.; Fryns, J.P.; Vermeesch, J.R. Piecing together the problems in diagnosing low-level chromosomal mosaicism. Genome Med. 2010, 2, 47. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Interphase chromosomes of the human brain: The biological and clinical meaning of neural aneuploidy. In Human Interphase Chromosomes; Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y., Eds.; Springer: New York, NY, USA, 2013; pp. 53–83. [Google Scholar]
- Yadav, V.K.; DeGregori, J.; De, S. The landscape of somatic mutations in protein coding genes in apparently benign human tissues carries signatures of relaxed purifying selection. Nucleic Acids Res. 2016, 44, 2075–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Human molecular neurocytogenetics. Curr. Genet. Med. Rep. 2018, 6, 155–164. [Google Scholar] [CrossRef]
- Rohrback, S.; Siddoway, B.; Liu, C.S.; Chun, J. Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 2018, 78, 1026–1048. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.J.; Dockery, H.E.; Fitzgerald, P.H.; Parfitt, R.G.; Romain, D.R.; Scobie, N.; Shaw, R.L.; Tumewu, P.; Watt, A.J. Mosaicism with a normal cell line and an autosomal structural rearrangement. J. Med. Genet. 1994, 31, 108–114. [Google Scholar] [CrossRef]
- Robinson, W.P.; Binkert, F.; Bernasconi, F.; Lorda-Sanchez, I.; Werder, E.A.; Schinzel, A.A. Molecular studies of chromosomal mosaicism: Relative frequency of chromosome gain or loss and possible role of cell selection. Am. J. Hum. Genet. 1995, 56, 444–451. [Google Scholar]
- Yurov, Y.B.; Vorsanova, S.G.; Solov’ev, I.V.; Iourov, I.Y. Instability of chromosomes in human nerve cells (normal and with neuromental diseases). Russ. J. Genet. 2010, 46, 1194–1196. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef]
- Knouse, K.A.; Wu, J.; Whittaker, C.A.; Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 13409–13414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bos, H.; Spierings, D.C.; Taudt, A.S.; Bakker, B.; Porubský, D.; Falconer, E.; Novoa, C.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 2016, 17, 116. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Human interphase chromosomes: A review of available molecular cytogenetic technologies. Mol. Cytogenet. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. (Eds.) Human Interphase Chromosomes: Biomedical Aspects; Springer: New York, NY, USA, 2013. [Google Scholar]
- Bakker, B.; van den Bos, H.; Lansdorp, P.M.; Foijer, F. How to count chromosomes in a cell: An overview of current and novel technologies. BioEssays 2015, 37, 570–577. [Google Scholar] [CrossRef]
- Goldman, S.L.; MacKay, M.J.; Afshinnekoo, E.; Melnick, A.; Wu, S.; Mason, C.E. The impact of heterogeneity on single-cell sequencing. Front. Genet. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Yurov, Y.B.; Vorsanova, S.G. Recent patents on molecular cytogenetics. Recent Pat. DNA Gene Seq. 2008, 2, 6–15. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Single cell genomics of the brain: Focus on neuronal diversity and neuropsychiatric diseases. Curr. Genom. 2012, 13, 477–488. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Technological solutions in human interphase cytogenetics. In Human Interphase Chromosomes; Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y., Eds.; Springer: New York, NY, USA, 2013; pp. 179–203. [Google Scholar]
- Rodríguez-Santiago, B.; Malats, N.; Rothman, N.; Armengol, L.; Garcia-Closas, M.; Kogevinas, M.; Villa, O.; Hutchinson, A.; Earl, J.; Marenne, G.; et al. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am. J. Hum. Genet. 2010, 87, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Voinova, V.Y.; Yurov, I.Y.; Kurinnaya, O.S.; Demidova, I.A.; Yurov, Y.B. Cytogenetic, molecular-cytogenetic, and clinical-genealogical studies of the mothers of children with autism: A search for familial genetic markers for autistic disorders. Neurosci. Behav. Physiol. 2010, 40, 745–756. [Google Scholar] [CrossRef]
- Veenma, D.; Brosens, E.; de Jong, E.; van de Ven, C.; Meeussen, C.; Cohen-Overbeek, T.; Boter, M.; Eussen, H.; Douben, H.; Tibboel, D.; et al. Copy number detection in discordant monozygotic twins of Congenital Diaphragmatic Hernia (CDH) and Esophageal Atresia (EA) cohorts. Eur. J. Hum. Genet. 2012, 20, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Yuan, B.; Robberecht, C.; Pfundt, R.; Szafranski, P.; McEntagart, M.E.; Nagamani, S.C.; Erez, A.; Bartnik, M.; Wiśniowiecka-Kowalnik, B.; et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 2014, 95, 173–182. [Google Scholar] [CrossRef]
- Bonaglia, M.C.; Kurtas, N.E.; Errichiello, E.; Bertuzzo, S.; Beri, S.; Mehrjouy, M.M.; Provenzano, A.; Vergani, D.; Pecile, V.; Novara, F.; et al. De novo unbalanced translocations have a complex history/aetiology. Hum. Genet. 2018, 137, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Monakhov, V.V.; Soloviev, I.V.; Yurov, Y.B. Dynamic mosaicism manifesting as loss, gain and rearrangement of an isodicentric Y chromosome in a male child with growth retardation and abnormal external genitalia. Cytogenet. Genome Res. 2008, 121, 302–306. [Google Scholar] [CrossRef]
- Erickson, R.P. Somatic gene mutation and human disease other than cancer: An update. Mutat. Res. 2010, 705, 96–106. [Google Scholar] [CrossRef]
- Conlin, L.K.; Kramer, W.; Hutchinson, A.L.; Li, X.; Riethman, H.; Hakonarson, H.; Mulley, J.C.; Scheffer, I.E.; Berkovic, S.F.; Hosain, S.A.; et al. Molecular analysis of ring chromosome 20 syndrome reveals two distinct groups of patients. J. Med. Genet. 2011, 48, 1–9. [Google Scholar] [CrossRef]
- Kim, J.W.; Park, S.Y.; Ryu, H.M.; Lee, D.E.; Lee, B.Y.; Kim, S.Y.; Park, Y.S.; Lee, H.S.; Seo, J.T. Molecular and clinical characteristics of 26 cases with structural Y chromosome aberrations. Cytogenet. Genome Res. 2012, 136, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Chai, H.; Shu, W.; Li, P. Human ring chromosome registry for cases in the Chinese population: Re-emphasizing cytogenomic and clinical heterogeneity and reviewing diagnostic and treatment strategies. Mol. Cytogenet. 2018, 11, 19. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Iourov, I.Y.; Voinova-Ulas, V.Y.; Weise, A.; Monakhov, V.V.; Kolotii, A.D.; Soloviev, I.V.; Novikov, P.V.; Yurov, Y.B.; Liehr, T. Partial monosomy 7q34-qter and 21pter-q22.13 due to cryptic unbalanced translocation t(7;21) but not monosomy of the whole chromosome 21: A case report plus review of the literature. Mol. Cytogenet. 2008, 1, 13. [Google Scholar] [CrossRef]
- Hook, E.B.; Warburton, D. Turner syndrome revisited: Review of new data supports the hypothesis that all viable 45,X cases are cryptic mosaics with a rescue cell line, implying an origin by mitotic loss. Hum. Genet. 2014, 133, 417–424. [Google Scholar] [CrossRef]
- Jackson-Cook, C. Constitutional and acquired autosomal aneuploidy. Clin. Lab. Med. 2011, 31, 481–511. [Google Scholar] [CrossRef] [PubMed]
- Dumanski, J.P.; Piotrowski, A. Structural genetic variation in the context of somatic mosaicism. Methods Mol. Biol. 2012, 838, 249–272. [Google Scholar] [PubMed]
- Howard, P.J.; Cramp, C.E.; Fryer, A.E. Trisomy 1 mosaicism only detected on a direct chromosome preparation in a neonate. Clin. Genet. 1995, 48, 313–316. [Google Scholar] [CrossRef]
- Wolstenholme, J. Confined placental mosaicism for trisomies 2, 3, 7, 8, 9, 16, and 22: Their incidence, likely origins, and mechanisms for cell lineage compartmentalization. Prenat. Diagn. 1996, 16, 511–524. [Google Scholar] [CrossRef]
- Wan, J.; Li, R.; Zhang, Y.; Jing, X.; Yu, Q.; Li, F.; Li, Y.; Zhang, L.; Yi, C.; Li, J.; et al. Pregnancy outcome of autosomal aneuploidies other than common trisomies detected by noninvasive prenatal testing in routine clinical practice. Prenat. Diagn. 2018, 38, 849–857. [Google Scholar] [CrossRef]
- Gupta, S.; Shah, S.; Mcgaw, A.; Mercado, T.; Zaslav, A.L.; Tegay, D. Trisomy 2 mosaicism in hypomelanosis of Ito. Am. J. Med. Genet. A 2007, 143, 2466–2468. [Google Scholar] [CrossRef] [PubMed]
- Prontera, P.; Stangoni, G.; Ardisia, C.; Rogaia, D.; Mencarelli, A.; Donti, E. Trisomy 2 mosaicism with caudal dysgenesis, Hirschsprung disease, and micro-anophthalmia. Am. J. Med. Genet. A 2011, 155, 928–930. [Google Scholar] [CrossRef] [PubMed]
- Kekis, M.; Hashimoto, S.; Deeg, C.; Calloway, I.; McKinney, A.; Shuss, C.; Hickey, S.; Astbury, C. A case of constitutional trisomy 3 mosaicism in a teenage patient with mild phenotype. Eur. J. Med. Genet. 2016, 59, 569–572. [Google Scholar] [CrossRef]
- Yang, Y.J.; Yao, X.; Guo, J.; Zhao, L.; Tu, M.; Qiou, J.; Zhao, R.; Luo, Y.; Zhu, Y.M. Trisomy 3 mosaicism in a 5-year-old boy with multiple anomalies: A very rare case. Am. J. Med. Genet. A 2016, 170, 1590–1594. [Google Scholar] [CrossRef]
- Bouman, A.; van der Kevie-Kersemaekers, A.M.; Huijsdens-van Amsterdam, K.; Dahhan, N.; Knegt, L.; Vansenne, F.; Cobben, J.M. Trisomy 4 mosaicism: Delineation of the phenotype. Am. J. Med. Genet. A 2016, 170, 1040–1045. [Google Scholar] [CrossRef]
- Reittinger, A.M.; Helm, B.M.; Boles, D.J.; Gadi, I.K.; Schrier Vergano, S.A. A prenatal diagnosis of mosaic trisomy 5 reveals a postnatal complete uniparental disomy of chromosome 5 with multiple congenital anomalies. Am. J. Med. Genet. A 2017, 173, 2528–2533. [Google Scholar] [CrossRef]
- Warburton, D. Trisomy 7 mosaic: Prognosis after prenatal diagnosis. Prenat. Diagn. 2002, 22, 1239–1240. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Holder-Espinasse, M.; Duban-Bedu, B.; Bouquillon, S.; Boute-Benejean, O.; Bazin, A.; Rouland, V.; Manouvrier-Hanu, S.; Delobel, B. Trisomy 7 mosaicism prenatally misdiagnosed and maternal uniparental disomy in a child with pigmentary mosaicism and Russell-Silver syndrome. Clin. Genet. 2012, 81, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Abdelhedi, F.; El Khattabi, L.; Cuisset, L.; Tsatsaris, V.; Viot, G.; Druart, L.; Lebbar, A.; Dupont, J.M. Neonatal Silver-Russell syndrome with maternal uniparental heterodisomy, trisomy 7 mosaicism, and dysplasia of the cerebellum. Am. J. Clin. Pathol. 2014, 142, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Hale, N.E.; Keane, J.F., Jr. Piecing together a picture of trisomy 8 mosaicism syndrome. J. Am. Osteopath Assoc. 2010, 110, 21–23. [Google Scholar] [PubMed]
- Cassina, M.; Calò, A.; Salviati, L.; Alghisi, A.; Montaldi, A.; Clementi, M. Prenatal detection of trisomy 8 mosaicism: Pregnancy outcome and follow up of a series of 17 consecutive cases. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 221, 23–27. [Google Scholar] [CrossRef]
- Bruns, D.A.; Campbell, E. Twenty-five additional cases of trisomy 9 mosaic: Birth information, medical conditions, and developmental status. Am. J. Med. Genet. A 2015, 167, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Hahnemann, J.M.; Nir, M.; Friberg, M.; Engel, U.; Bugge, M. Trisomy 10 mosaicism and maternal uniparental disomy 10 in a liveborn infant with severe congenital malformations. Am. J. Med. Genet. A 2005, 138, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.; Peres, L.C.; Pelly, D. Mosaic trisomy 11 in a fetus with bilateral renal agenesis: Co-incidence or new association? Clin. Dysmorphol. 2011, 20, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Zunich, J.; Openshaw, A.; Toydemir, R.M. Clinical features of trisomy 12 mosaicism-Report and review. Am. J. Med. Genet. A 2017, 173, 1681–1686. [Google Scholar] [CrossRef] [PubMed]
- Karaman, B.; Kayserili, H.; Ghanbari, A.; Uyguner, Z.O.; Toksoy, G.; Altunoglu, U.; Basaran, S. Pallister-Killian syndrome: Clinical, cytogenetic and molecular findings in 15 cases. Mol. Cytogenet. 2018, 11, 45. [Google Scholar] [CrossRef]
- Thakur, S.; Gupta, R.; Tiwari, B.; Singh, N.; Saxena, K.K. Pallister-Killian syndrome: Review of fetal phenotype. Clin. Genet. 2019, 95, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.F.; Hou, J.W. Variable expressivity in Patau syndrome is not all related to trisomy 13 mosaicism. Am. J. Med. Genet. A 2007, 143, 1739–1748. [Google Scholar] [CrossRef]
- Griffith, C.B.; Vance, G.H.; Weaver, D.D. Phenotypic variability in trisomy 13 mosaicism: Two new patients and literature review. Am. J. Med. Genet. A 2009, 9149, 1346–1358. [Google Scholar] [CrossRef]
- Jinawath, N.; Zambrano, R.; Wohler, E.; Palmquist, M.K.; Hoover-Fong, J.; Hamosh, A.; Batista, D.A. Mosaic trisomy 13: Understanding origin using SNP array. J. Med. Genet. 2011, 148, 323–326. [Google Scholar] [CrossRef]
- Salas-Labadía, C.; Lieberman, E.; Cruz-Alcívar, R.; Navarrete-Meneses, P.; Gómez, S.; Cantú-Reyna, C.; Buiting, K.; Durán-McKinster, C.; Pérez-Vera, P. Partial and complete trisomy 14 mosaicism: Clinical follow-up, cytogenetic and molecular analysis. Mol. Cytogenet. 2014, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qin, H.; Wang, J.; OuYang, L.; Luo, S.; Fu, C.; Fan, X.; Su, J.; Chen, R.; Xie, B.; et al. Maternal uniparental disomy 14 and mosaic trisomy 14 in a Chinese boy with moderate to severe intellectual disability. Mol. Cytogenet. 2016, 91, 66. [Google Scholar] [CrossRef] [PubMed]
- McPadden, J.; Helm, B.M.; Spangler, B.B.; Ross, L.P.; Boles, D.B.; Schrier Vergano, S.A. Mosaic trisomy 15 in a liveborn infant. Am. J. Med. Genet. A 2015, 167, 821–825. [Google Scholar] [CrossRef]
- Eggermann, T.; Soellner, L.; Buiting, K.; Kotzot, D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol. Med. 2015, 21, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Langlois, S.; Yong, P.J.; Yong, S.L.; Barrett, I.; Kalousek, D.K.; Miny, P.; Exeler, R.; Morris, K.; Robinson, W.P. Postnatal follow-up of prenatally diagnosed trisomy 16 mosaicism. Prenat. Diagn. 2006, 26, 548–558. [Google Scholar] [CrossRef]
- Sparks, T.N.; Thao, K.; Norton, M.E. Mosaic trisomy 16: What are the obstetric and long-term childhood outcomes? Genet. Med. 2017, 19, 1164–1170. [Google Scholar] [CrossRef]
- De Vries, F.A.; Govaerts, L.C.; Knijnenburg, J.; Knapen, M.F.; Oudesluijs, G.G.; Lont, D.; Noomen, P.; de Graaff, K.; Srebniak, M.I.; Van Opstal, D. Another rare prenatal case of post-zygotic mosaic trisomy 17. Am. J. Med. Genet. A 2013, 161, 1196–1199. [Google Scholar] [CrossRef]
- Baltensperger, A.; Haischer, G.; Rohena, L. Rare case of live born with confirmed mosaic trisomy 17 and review of the literature. Clin. Case Rep. 2016, 4, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Banka, S.; Metcalfe, K.; Clayton-Smith, J. Trisomy 18 mosaicism: Report of two cases. World J. Pediatr. 2013, 9, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Fitas, A.L.; Paiva, M.; Cordeiro, A.I.; Nunes, L.; Cordeiro-Ferreira, G. Mosaic trisomy 18 in a five-month-old infant. Case Rep. Pediatr. 2013, 2013, 929861. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.E.; Rosella, L.C.; Mahant, S.; Guttmann, A. Survival and surgical interventions for children with trisomy 13 and 18. JAMA 2016, 316, 420–428. [Google Scholar] [CrossRef]
- Chen, H.; Yu, C.W.; Wood, M.J.; Landry, K. Mosaic trisomy 19 syndrome. Ann. Genet. 1981, 24, 32–33. [Google Scholar]
- Wallerstein, R.; Twersky, S.; Layman, P.; Kernaghan, L.; Aviv, H.; Pedro, H.F.; Pletcher, B. Long term follow-up of developmental delay in a child with prenatally-diagnosed trisomy 20 mosaicism. Am. J. Med. Genet. A 2005, 137, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.J.H.; Bird, L.M.; Dell’Aquilla, M.; Jones, M.C. Expanding the phenotype of mosaic trisomy 20. Am. J. Med. Genet. A 2008, 146, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Roizen, N.J.; Patterson, D. Down’s syndrome. Lancet 2003, 361, 1281–1289. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Iourov, I.Y.; Beresheva, A.K.; Demidova, I.A.; Monakhov, V.V.; Kravets, V.S.; Bartseva, O.B.; Goyko, E.A.; Soloviev, I.V.; Yurov, Y.B. Non-disjunction of chromosome 21, alphoid DNA variation, and sociogenetic features of Down syndrome. Tsitol. Genet. 2005, 39, 30–36. [Google Scholar]
- Hultén, M.A.; Jonasson, J.; Nordgren, A.; Iwarsson, E. Germinal and Somatic Trisomy 21 mosaicism: How common is it, what are the implications for individual carriers and how does it come about? Curr. Genom. 2010, 11, 409–419. [Google Scholar] [CrossRef]
- Papavassiliou, P.; Charalsawadi, C.; Rafferty, K.; Jackson-Cook, C. Mosaicism for trisomy 21: A review. Am. J. Med. Genet. A 2015, 167, 26–39. [Google Scholar] [CrossRef]
- Antonarakis, S.E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 2017, 18, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, S.; Baron, X.; Jacquemont, M.L.; Cuillier, F.; Cartault, F. Mosaic trisomy 22: Five new cases with variable outcomes. Implications for genetic counselling and clinical management. Prenat. Diagn. 2010, 30, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Abdelgadir, D.; Nowaczyk, M.J.; Li, C. Trisomy 22 mosaicism and normal developmental outcome: Report of two patients and review of the literature. Am. J. Med. Genet. A 2013, 161, 1126–1131. [Google Scholar] [CrossRef]
- Tinkle, B.T.; Walker, M.E.; Blough-Pfau, R.I.; Saal, H.M.; Hopkin, R.J. Unexpected survival in a case of prenatally diagnosed non-mosaic trisomy 22: Clinical report and review of the natural history. Am. J. Med. Genet. A 2003, 118, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Skuse, D.; Printzlau, F.; Wolstencroft, J. Sex chromosome aneuploidies. Handb. Clin. Neurol. 2018, 147, 355–376. [Google Scholar]
- Sybert, V.P.; McCauley, E. Turner’s syndrome. N. Eng. J. Med. 2004, 351, 1227–1238. [Google Scholar] [CrossRef]
- Tuke, M.A.; Ruth, K.S.; Wood, A.R.; Beaumont, R.N.; Tyrrell, J.; Jones, S.E.; Yaghootkar, H.; Turner, C.L.S.; Donohoe, M.E.; Brooke, A.M.; et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Iurov, I.; Vorsanova, S.G.; Iurov, I. Chromosome abnormalities in schizophrenia. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova 2006, 106, 75–82. [Google Scholar]
- Green, T.; Flash, S.; Reiss, A.L. Sex differences in psychiatric disorders: What we can learn from sex chromosome aneuploidies. Neuropsychopharmacology 2019, 44, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, I.; Lleo, A.; Gershwin, M.E.; Invernizzi, P. The X chromosome and immune associated genes. J. Autoimmun. 2012, 38, J187–J192. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Guo, H.R. Mechanism of recurrent spontaneous abortions in women with mosaicism of X-chromosome aneuploidies. Fertil. Steril. 2004, 82, 1594–1601. [Google Scholar] [CrossRef]
- Gersak, K.; Veble, A. Low-level X chromosome mosaicism in women with sporadic premature ovarian failure. Reprod. Biomed. Online 2011, 22, 399–403. [Google Scholar] [CrossRef]
- Russell, L.M.; Strike, P.; Browne, C.E.; Jacobs, P.A. X chromosome loss and ageing. Cytogenet. Genome Res. 2007, 116, 181–185. [Google Scholar] [CrossRef]
- Guc-Scekic, M.; Milasin, J.; Stevanovic, M.; Stojanov, L.J.; Djordjevic, M. Tetraploidy in a 26-month-old girl (cytogenetic and molecular studies). Clin. Genet. 2002, 61, 62–65. [Google Scholar] [CrossRef]
- Rosenbusch, B.; Schneider, M. A brief look at the origin of tetraploidy. Cytogenet. Genome Res. 2004, 107, 128–131. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Iourov, I.Y.; Kolotii, A.D.; Beresheva, A.K.; Demidova, I.A.; Kurinnaya, O.S.; Kravets, V.S.; Monakhov, V.V.; Soloviev, I.V.; Yurov, Y.B. Chromosomal mosaicism in spontaneous abortions: Analysis of 650 cases. Russ. J. Genet. 2010, 46, 1197–1200. [Google Scholar] [CrossRef]
- Gentric, G.; Desdouets, C. Polyploidization in liver tissue. Am. J. Pathol. 2014, 184, 322–331. [Google Scholar] [CrossRef]
- Chow, H.M.; Herrup, K. Genomic integrity and the ageing brain. Nat. Rev. Neurosci. 2015, 16, 672–684. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Genomic landscape of the Alzheimer’s disease brain: Chromosome instability—aneuploidy, but not tetraploidy—mediates neurodegeneration. Neurodegener. Dis. 2011, 8, 35–37. [Google Scholar] [CrossRef]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T. Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Regan, S. A systems biology perspective on molecular cytogenetics. Curr. Bioinform. 2017, 12, 4–10. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Yurov, Y.B.; Iourov, I.Y. Neurogenomic pathway of autism spectrum disorders: Linking germline and somatic mutations to genetic-environmental interactions. Curr. Bioinform. 2017, 12, 19–26. [Google Scholar] [CrossRef]
- Heng, H.H.; Horne, S.D.; Chaudhry, S.; Regan, S.M.; Liu, G.; Abdallah, B.Y.; Ye, C.J. A postgenomic perspective on molecular cytogenetics. Curr. Genom. 2018, 19, 227–239. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Pathway-based classification of genetic diseases. Mol. Cytogenet. 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H. Genome Chaos; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Abdallah, B.Y.; Horne, S.D.; Stevens, J.B.; Liu, G.; Ying, A.Y.; Vanderhyden, B.; Krawetz, S.A.; Gorelick, R.; Heng, H.H. Single cell heterogeneity: Why unstable genomes are incompatible with average profiles. Cell Cycle 2013, 12, 3640–3649. [Google Scholar] [CrossRef] [PubMed]
- Junker, J.P.; van Oudenaarden, A. Every cell is special: Genome-wide studies add a new dimension to single-cell biology. Cell 2014, 157, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Komin, N.; Skupin, A. How to address cellular heterogeneity by distribution biology. Curr. Opin. Syst. Biol. 2017, 3, 154–160. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H. Phenotypes and genotypes of the chromosomal instability syndromes. Transl. Pediatr. 2016, 5, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frias, S.; Ramos, S.; Salas, C.; Molina, B.; Sánchez, S.; Rivera-Luna, R. Nonclonal chromosome aberrations and genome chaos in somatic and germ cells from patients and survivors of hodgkin lymphoma. Genes 2019, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.H.; Bremer, S.W.; Stevens, J.B.; Horne, S.D.; Liu, G.; Abdallah, B.Y.; Karen, J.Y.; Christine, J.Y. Chromosomal instability (CIN): What it is and why it is crucial to cancer evolution. Cancer Metastasis Rev. 2013, 32, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Hirota, T. Chromosomal instability: A common feature and a therapeutic target of cancer. Biochim. Biophys. Acta 2016, 1866, 64–75. [Google Scholar] [CrossRef]
- Simonetti, G.; Bruno, S.; Padella, A.; Tenti, E.; Martinelli, G. Aneuploidy: Cancer strength or vulnerability? Int. J. Cancer 2019, 144, 8–25. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Zelenova, M.A.; Korostelev, S.A.; Yurov, Y.B. Genomic copy number variation affecting genes involved in the cell cycle pathway: Implications for somatic mosaicism. Int. J. Genomics 2015, 2015, 757680. [Google Scholar] [CrossRef]
- Li, Y.; Agarwal, P. A pathway-based view of human diseases and disease relationships. PLoS ONE 2009, 4, e4346. [Google Scholar] [CrossRef]
- Putnam, C.D.; Allen-Soltero, S.R.; Martinez, S.L.; Chan, J.E.; Hayes, T.K.; Kolodner, R.D. Bioinformatic identification of genes suppressing genome instability. Proc. Natl. Acad. Sci. USA 2012, 109, E3251–E3259. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. In silico molecular cytogenetics: A bioinformatic approach to prioritization of candidate genes and copy number variations for basic and clinical genome research. Mol. Cytogenet. 2014, 7, 98. [Google Scholar] [CrossRef]
- Song, L.; Bhuvaneshwar, K.; Wang, Y.; Feng, Y.; Shih, I.M.; Madhavan, S.; Gusev, Y. CINdex: A Bioconductor package for analysis of chromosome instability in DNA copy number data. Cancer Inform. 2017, 16, 1176935117746637. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. Network-based classification of molecular cytogenetic data. Curr. Bioinform. 2017, 12, 27–33. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Vorsanova, S.G.; Demidova, I.A.; Aliamovskaia, G.A.; Keshishian, E.S.; Yurov, Y.B. 5p13.3p13.2 duplication associated with developmental delay, congenital malformations and chromosome instability manifested as low-level aneuploidy. Springerplus 2015, 4, 616. [Google Scholar] [CrossRef]
- Dierssen, M.; Herault, Y.; Estivill, X. Aneuploidy: From a physiological mechanism of variance to Down syndrome. Physiol. Rev. 2009, 89, 887–920. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Boveri revisited: Chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Machiela, M.J. Mosaicism, aging and cancer. Curr. Opin. Oncol. 2019, 31, 108–113. [Google Scholar] [CrossRef]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef]
- Schneider, G.; Schmidt-Supprian, M.; Rad, R.; Saur, D. Tissue-specific tumorigenesis: Context matters. Nat. Rev. Cancer 2017, 17, 239–253. [Google Scholar] [CrossRef]
- Risques, R.A.; Kennedy, S.R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef]
- Fortunato, A.; Boddy, A.; Mallo, D.; Aktipis, A.; Maley, C.C.; Pepper, J.W. Natural selection in cancer biology: From molecular snowflakes to trait hallmarks. Cold Spring Harb. Perspect. Med. 2017, 7, a029652. [Google Scholar] [CrossRef]
- Van Jaarsveld, R.H.; Kops, G.J.P.L. Difference makers: Chromosomal instability versus aneuploidy in cancer. Trends Cancer 2016, 2, 561–571. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Yurov, Y.B. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. 2009, 18, 2656–2669. [Google Scholar] [CrossRef] [Green Version]
- Arendt, T.; Brückner, M.K.; Mosch, B.; Lösche, A. Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 2010, 177, 15–20. [Google Scholar] [CrossRef]
- Driver, J.A. Understanding the link between cancer and neurodegeneration. J. Geriatr. Oncol. 2012, 3, 58–67. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vostrikov, V.M.; Vorsanova, S.G.; Monakhov, V.V.; Iourov, I.Y. Multicolor fluorescent in situ hybridization on post-mortem brain in schizophrenia as an approach for identification of low-level chromosomal aneuploidy in neuropsychiatric diseases. Brain Dev. 2001, 23 (Suppl. 1), S186–S190. [Google Scholar] [CrossRef]
- Bushman, D.M.; Chun, J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin. Cell Dev. Biol. 2013, 24, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Somatic cell genomics of brain disorders: A new opportunity to clarify genetic-environmental interactions. Cytogenet. Genome Res. 2013, 139, 181–188. [Google Scholar] [CrossRef]
- Paquola, A.C.M.; Erwin, J.A.; Gage, F.H. Insights into the role of somatic mosaicism in the brain. Curr. Opin. Syst. Biol. 2017, 1, 90–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, M.J.; Moran, J.V.; Abyzov, A.; Akbarian, S.; Bae, T.; Cortes-Ciriano, I.; Erwin, J.A.; Fasching, L.; Flasch, D.A.; Freed, D.; et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 2017, 356, eaal1641. [Google Scholar] [CrossRef] [Green Version]
- Rodin, R.E.; Walsh, C.A. Somatic mutation in pediatric neurological diseases. Pediatr. Neurol. 2018, 87, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G.; Yurov, I.Y.; Demidova, I.A.; Voinova-Ulas, V.Y.; Kravets, V.S.; Solov’ev, I.V.; Gorbachevskaya, N.L.; Yurov, Y.B. Variability in the heterochromatin regions of the chromosomes and chromosomal anomalies in children with autism: Identification of genetic markers of autistic spectrum disorders. Neurosci. Behav. Physiol. 2007, 37, 553–558. [Google Scholar] [CrossRef]
- Dou, Y.; Yang, X.; Li, Z.; Wang, S.; Zhang, Z.; Ye, A.Y.; Yan, L.; Yang, C.; Wu, Q.; Li, J.; et al. Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum. Mutat. 2017, 38, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Yurov, Y.B.; Vorsanova, S.G. Mosaic X chromosome aneuploidy can help to explain the male-to-female ratio in autism. Med. Hypotheses 2008, 70, 456. [Google Scholar] [CrossRef]
- Rivet, T.T.; Matson, J.L. Review of gender differences in core symptomatology in autism spectrum disorders. Res. Autism. Spectr. Disord. 2011, 5, 957–976. [Google Scholar] [CrossRef]
- Iourov, I.; Vorsanova, S.; Liehr, T.; Zelenova, M.; Kurinnaia, O.; Vasin, K.; Yurov, Y. Chromothripsis as a mechanism driving genomic instability mediating brain diseases. Mol. Cytogenet. 2017, 10 (Suppl. 1), 20. [Google Scholar]
- Vijayakumar, N.T.; Judy, M.V. Autism spectrum disorders: Integration of the genome, transcriptome and the environment. J. Neurol. Sci. 2016, 364, 167–176. [Google Scholar] [CrossRef]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A Review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Demidova, I.A.; Kolotii, A.D.; Soloviev, I.V.; Iourov, I.Y. Mosaic brain aneuploidy in mental illnesses: An association of low-level post-zygotic aneuploidy with schizophrenia and comorbid psychiatric disorders. Curr. Genom. 2018, 19, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G.; Demidova, I.A.; Kravetz, V.S.; Beresheva, A.K.; Kolotii, A.D.; Monakchov, V.V.; Uranova, N.A.; Vostrikov, V.M.; et al. The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr. Res. 2008, 98, 139–147. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Demidova, I.A.; Kravets, V.S.; Vostrikov, V.M.; Soloviev, I.V.; Uranova, N.A.; Iourov, I.Y. Genomic instability in the brain: Chromosomal mosaicism in schizophrenia. Zhurnal Nevrologii i Psikhiatrii Imeni SS Korsakova 2016, 116, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Watanabe, Y.; Someya, T.; Araki, K.; Shibuya, M.; Niizato, K.; Oshima, K.; Kunii, Y.; Yabe, H.; Matsumoto, J.; et al. Assessment of copy number variations in the brain genome of schizophrenia patients. Mol. Cytogenet. 2015, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Leija-Salazar, M.; Piette, C.; Proukakis, C. Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 2018, 44, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.E.; Yang, Y.; Halliday, G.M. Region- and Cell-specific aneuploidy in brain aging and neurodegeneration. Neuroscience 2018, 374, 326–334. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Bohr, V.A.; Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 2011, 94, 166–200. [Google Scholar] [CrossRef]
- Coppede, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Ataxia telangiectasia paradox can be explained by chromosome instability at the subtissue level. Med. Hypotheses 2007, 68, 716. [Google Scholar] [CrossRef]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet. J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol. Dis. 2009, 34, 212–220. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.J.; Kaushal, D.; Yang, A.H.; Kingsbury, M.A.; Rehen, S.K.; Treuner, K.; Helton, R.; Annas, E.G.; Chun, J.; Barlow, C. Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J. Neurosci. 2004, 24, 8090–8096. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Iourov, I.Y.; Vorsanova, S.G. Neurodegeneration mediated by chromosome instability suggests changes in strategy for therapy development in ataxia-telangiectasia. Med. Hypotheses 2009, 73, 1075–1076. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Xu, B. The versatile functions of ATM kinase. Biomed. J. 2014, 37, 3–9. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Developmental neural chromosome instability as a possible cause of childhood brain cancers. Med. Hypotheses 2009, 72, 615–616. [Google Scholar] [CrossRef]
- Potter, H.; Granic, A.; Caneus, J. Role of trisomy 21 mosaicism in sporadic and familial Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Iourov, I.Y. X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 2014, 7, 20. [Google Scholar] [CrossRef]
- Arendt, T.; Brückner, M.K.; Lösche, A. Regional mosaic genomic heterogeneity in the elderly and in Alzheimer’s disease as a correlate of neuronal vulnerability. Acta Neuropathol. 2015, 130, 501–510. [Google Scholar] [CrossRef]
- Bushman, D.M.; Kaeser, G.E.; Siddoway, B.; Westra, J.W.; Rivera, R.R.; Rehen, S.K.; Yung, Y.C.; Chun, J. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife 2015, 4, e05116. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Siddoway, B.; Kaeser, G.E.; Segota, I.; Rivera, R.; Romanow, W.J.; Liu, C.S.; Park, C.; Kennedy, G.; Long, T.; et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 2018, 563, 639–645. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. The DNA replication stress hypothesis of Alzheimer’s disease. Sci. World J. 2011, 11, 2602–2612. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.; Spremo-Potparevic, B.; Zivkovic, L.; Isenovic, E.R.; Arendt, T. Cohesion and the aneuploid phenotype in Alzheimer’s disease: A tale of genome instability. Neurosci. Biobehav. Rev. 2015, 55, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.; Ueberham, U. Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 2017, 143, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Granic, A.; Potter, H. Mitotic spindle defects and chromosome mis-segregation induced by LDL/cholesterol-implications for Niemann-Pick C1, Alzheimer’s disease, and atherosclerosis. PLoS ONE 2013, 8, e60718. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shepherd, C.; Halliday, G. Aneuploidy in Lewy body diseases. Neurobiol. Aging 2015, 36, 1253–1260. [Google Scholar] [CrossRef]
- Caneus, J.; Granic, A.; Rademakers, R.; Dickson, D.W.; Coughlan, C.M.; Chial, H.J.; Potter, H. Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol. Biol. Cell 2018, 29, 575–586. [Google Scholar] [CrossRef]
- Peterson, S.E.; Yang, A.H.; Bushman, D.M.; Westra, J.W.; Yung, Y.C.; Barral, S.; Mutoh, T.; Rehen, S.K.; Chun, J. Aneuploid cells are differentially susceptible to caspase-mediated death during embryonic cerebral cortical development. J. Neurosci. 2012, 32, 16213–16222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charney, E. Behavior genetics and postgenomics. Behav. Brain Sci. 2012, 35, 331–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; Ye, C.J.; Chowdhury, S.K.; Abdallah, B.Y.; Horne, S.D.; Nichols, D.; Heng, H.H. Detecting chromosome condensation defects in gulf war illness patients. Curr. Genom. 2018, 19, 200–206. [Google Scholar] [CrossRef]
- Vorsanova, S.G.; Zelenova, M.A.; Yurov, Y.B.; Iourov, I.Y. Behavioral variability and somatic mosaicism: A cytogenomic hypothesis. Curr. Genom. 2018, 19, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Emerit, I.; Michelson, A.M. Chromosome instability in human and murine autoimmune disease: Anticlastogenic effect of superoxide dismutase. Acta Physiol. Scand. Suppl. 1980, 492, 59–65. [Google Scholar]
- Laish, I.; Mannasse-Green, B.; Hadary, R.; Konikoff, F.M.; Amiel, A.; Kitay-Cohen, Y. Aneuploidy and asynchronous replication in non-alcholic fatty liver disease and cryptogenic cirrhosis. Gene 2016, 593, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.H.; Moscato, Z.; Choate, K.A. Mosaicism in cutaneous disorders. Annu. Rev. Genet. 2017, 51, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, H.; Sato, K.; Habuta, M.; Fujita, H.; Bando, T. Congenital eye anomalies: More mosaic than thought? Congenit. Anom. 2018. [Google Scholar] [CrossRef]
- Astolfi, P.A.; Salamini, F.; Sgaramella, V. Are we genomic mosaics? Variations of the genome of somatic cells can contribute to diversify our phenotypes. Curr. Genom. 2010, 11, 379–386. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Mosaike im Gehirn des Menschen. Med. Genet. 2014, 26, 342–345. [Google Scholar] [CrossRef]
- Arendt, T.; Mosch, B.; Morawski, M. Neuronal aneuploidy in health and disease: A cytomic approach to understand the molecular individuality of neurons. Int. J. Mol. Sci. 2009, 10, 1609–1627. [Google Scholar] [CrossRef] [PubMed]
- Liehr, T. Fluorescence in situ Hybridization (FISH); Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Levy, B.; Burnside, R.D. Are all chromosome microarrays the same? What clinicians need to know? Prenat. Diagn. 2019, 39, 157–164. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Liehr, T.; Vorsanova, S.G.; Kolotii, A.D.; Yurov, Y.B. Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosome Res. 2006, 14, 223–229. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Liehr, T.; Vorsanova, S.G.; Yurov, Y.B. Interphase chromosome-specific multicolor banding (ICS-MCB): A new tool for analysis of interphase chromosomes in their integrity. Biomol. Eng. 2007, 24, 415–417. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Liehr, T.; Vorsanova, S.G.; Yurov, Y.B. Interphase chromosome-specific multicolor banding. In Human Interphase Chromosomes; Yurov, Y.B., Vorsanova, S.G., Iourov, I.Y., Eds.; Springer: New York, NY, USA, 2013; pp. 161–169. [Google Scholar]
- Lu, C.M.; Kwan, J.; Baumgartner, A.; Weier, J.F.; Wang, M.; Escudero, T.; Munné, S.; Zitzelsberger, H.F.; Weier, H.U. DNA probe pooling for rapid delineation of chromosomal breakpoints. J. Histochem. Cytochem. 2009, 57, 587–597. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Interphase FISH for detection of chromosomal mosaicism. In Fluorescence In Situ Hybridization (FISH); Springer Protocols Handbooks; Liehr, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 361–372. [Google Scholar]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. FISHing for unstable cellular genomes in the human brain. OBM Genet. 2019, 3, 11. [Google Scholar] [CrossRef]
- Wan, T.S.K. (Ed.) Cancer Cytogenetics: Methods and Protocols; Humana Press: New York, NY, USA, 2017. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Pellestor, F.; Yurov, Y.B. Brain tissue preparations for chromosomal PRINS labeling. Methods Mol. Biol. 2006, 334, 123–132. [Google Scholar]
- Yurov, Y.B.; Vorsanova, S.G.; Soloviev, I.V.; Ratnikov, A.M.; Iourov, I.Y. FISH-based assays for detecting genomic (chromosomal) mosaicism in human brain cells. Neuromethods 2017, 131, 27–41. [Google Scholar]
- Arnoldus, E.P.; Peters, A.C.; Bots, G.T.; Raap, A.K.; van der Ploeg, M. Somatic pairing of chromosome 1 centromeres in interphase nuclei of human cerebellum. Hum. Genet. 1989, 83, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y.; Soloviev, I.V.; Vorsanova, S.G.; Monakhov, V.V.; Yurov, Y.B. An approach for quantitative assessment of fluorescence in situ hybridization (FISH) signals for applied human molecular cytogenetics. J. Histochem. Cytochem. 2005, 53, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Iourov, I.Y. Quantitative fluorescence in situ hybridization (QFISH). Methods Mol. Biol. 2017, 1541, 143–149. [Google Scholar]
- Iourov, I.Y. Cytopostgenomics: What is it and how does it work? Curr. Genom. 2019, 20, 77–78. [Google Scholar]
- Vorsanova, S.G.; Yurov, Y.B.; Soloviev, I.V.; Kolotii, A.D.; Demidova, I.A.; Kravets, V.S.; Kurinnaia, O.S.; Zelenova, M.A.; Iourov, I.Y. FISH-based analysis of mosaic aneuploidy and chromosome instability for investigating molecular and cellular mechanisms of disease. OBM Genet. 2019, 3, 9. [Google Scholar] [CrossRef]
- Vijg, J.; Dong, X.; Milholland, B.; Zhang, L. Genome instability: A conserved mechanism of ageing? Essays Biochem. 2017, 61, 305–315. [Google Scholar] [CrossRef]
- Guttenbach, M.; Koschorz, B.; Bernthaler, U.; Grimm, T.; Schmid, M. Sex chromosome loss and aging: In situ hybridization studies on human interphase nuclei. Am. J. Hum. Genet. 1995, 57, 1143–1150. [Google Scholar]
- Spremo-Potparevic, B.; Bajic, V.; Perry, G.; Zivkovic, L. Alterations of the X chromosome in lymphocytes of Alzheimer’s disease patients. Curr. Alzheimer Res. 2015, 12, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Andriani, G.A.; Vijg, J.; Montagna, C. Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 2017, 161, 19–36. [Google Scholar] [CrossRef]
- Yurov, Y.B.; Vorsanova, S.G.; Iourov, I.Y. GIN’n’CIN hypothesis of brain aging: Deciphering the role of somatic genetic instabilities and neural aneuploidy during ontogeny. Mol. Cytogenet. 2009, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Faggioli, F.; Vijg, J.; Montagna, C. Chromosomal aneuploidy in the aging brain. Mech. Ageing Dev. 2011, 132, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faggioli, F.; Wang, T.; Vijg, J.; Montagna, C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum. Mol. Genet. 2012, 21, 5246–5253. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.G.; Morawski, M.; Brückner, M.K.; Mittag, A.; Tarnok, A.; Arendt, T. Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 2012, 11, 628–633. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Aida, J.; Izumiyama, N.; Ishikawa, N.; Fujiwara, M.; Poon, S.S.; Kondo, H.; Kammori, M.; Matsuura, M.; Sawabe, M.; et al. Chromosomal instability and telomere lengths of each chromosomal arm measured by Q-FISH in human fibroblast strains prior to replicative senescence. Mech. Ageing Dev. 2010, 131, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Vorsanova, S.G. Dynamics of changes in anomalous human cells during prolonged cultivation in the stationary phase. Trisomy 7 cells. Biulleten’eksperimental’noi Biologii i Meditsiny 1977, 83, 742–744. [Google Scholar]
- Mathon, N.F.; Lloyd, A.C. Cell senescence and cancer. Nat. Rev. Cancer 2001, 1, 203–213. [Google Scholar] [CrossRef]
- Oromendia, A.B.; Amon, A. Aneuploidy: Implications for protein homeostasis and disease. Dis. Model. Mech. 2014, 7, 15–20. [Google Scholar] [CrossRef]
- Bailey, K.J.; Maslov, A.Y.; Pruitt, S.C. Accumulation of mutations and somatic selection in aging neural stem/progenitor cells. Aging Cell 2004, 3, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villela, D.; Suemoto, C.K.; Leite, R.; Pasqualucci, C.A.; Grinberg, L.T.; Pearson, P.; Rosenberg, C. Increased DNA copy number variation mosaicism in elderly human brain. Neural Plast. 2018, 2018, 2406170. [Google Scholar] [CrossRef] [PubMed]
- Chronister, W.D.; Burbulis, I.E.; Wierman, M.B.; Wolpert, M.J.; Haakenson, M.F.; Smith, A.C.B.; Kleinman, J.E.; Hyde, T.M.; Weinberger, D.R.; Bekiranov, S.; et al. Neurons with complex karyotypes are rare in aged human neocortex. Cell Rep. 2019, 26, 825–835.e7. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B.; Kutsev, S.I. Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism. Genes 2019, 10, 379. https://doi.org/10.3390/genes10050379
Iourov IY, Vorsanova SG, Yurov YB, Kutsev SI. Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism. Genes. 2019; 10(5):379. https://doi.org/10.3390/genes10050379
Chicago/Turabian StyleIourov, Ivan Y., Svetlana G. Vorsanova, Yuri B. Yurov, and Sergei I. Kutsev. 2019. "Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism" Genes 10, no. 5: 379. https://doi.org/10.3390/genes10050379
APA StyleIourov, I. Y., Vorsanova, S. G., Yurov, Y. B., & Kutsev, S. I. (2019). Ontogenetic and Pathogenetic Views on Somatic Chromosomal Mosaicism. Genes, 10(5), 379. https://doi.org/10.3390/genes10050379