1. Introduction
Recent technological advances have allowed researchers to produce a large amount of genomic and transcriptomic data in a wide range of experimental systems. This wealth of data is usually uploaded in their raw format to public repositories, such as the Gene Expression Omnibus (GEO) Database (
https://www.ncbi.nlm.nih.gov/geo/), the European Bioinformatics Institute (EMBL-EBI) (
https://www.ebi.ac.uk/), the DNA Data Bank of Japan (DDBJ) (
https://www.ddbj.nig.ac.jp) and others, and can later be queried in a series of downstream applications and metanalyses by colleagues worldwide. While several tools exist for querying, retrieving and analyzing raw data deposited in public databases (e.g., a large collection of software packages distributed through the Bioconductor open source community;
www.bioconductor.org) [
1,
2], the generation of system-wide data still vastly outpaces its re-utilization in subsequent studies. For instance, the number of GEO dataset deposits is nearly ten times higher than the number of publications using previously deposited GEO datasets (59,331 vs. 7160 according to NCBI, GEO Citations Listings: deposits and third-party usage,
www.ncbi.nlm.nih.gov/geo/info/citations.html, accessed on 28 May 2019).
Such underutilization of data may result from a series of reasons. The scientific data management community has recently agreed that scientific data should be shared following four FAIR principles: Data should be Findable, Accessible, Interoperable and Reusable [
3]. While the public repositories listed above may ensure relatively well the findability and accessibility of raw datasets, their interoperability often depends on different extents of dataset pre-processing to ensure that they share formatting and relevant entities map correctly between files [
4]. In this regard, data reuse may be hindered by a pervasive lack of expertise in data management and processing by most experimental researchers [
5]. While bioinformaticians have developed various outstanding methods for accessing and reusing public datasets, these methods are often difficult to implement for experimental biologists without a programming background. In fact, it has been argued that such requirement for specialized training in the use of dedicated bioinformatics tools may contribute to the gap between data production and analysis and, somewhat ironically, slow down the publication of findings [
5].
Lastly, and perhaps most importantly, there exists an invaluable wealth of information in the form of processed data that is not easily ‘findable’ and/or ‘accessible’ and is therefore less amenable to automated analysis through bioinformatics (particularly by experimental life scientists with little or no programming experience). These include data that are often published as supplementary files and tables accompanying research publications, datasets that researchers may offer publicly through their own websites or upon request, data generated by online bioinformatics tools, or even extremely valuable data that may be unpublished but available to researchers in-house. Researchers with more advanced programming skills, or with easier access to collaborators who can provide bioinformatics support, can often generate simple ad hoc scripts that enable them to conduct a more exhaustive and careful integration of data from different sources and with heterogenous formatting. Unfortunately, however, many experimental biologists may be discouraged from attempting a more systematic integration of existing data given the time and effort involved in reformatting and integrating diverse datasets.
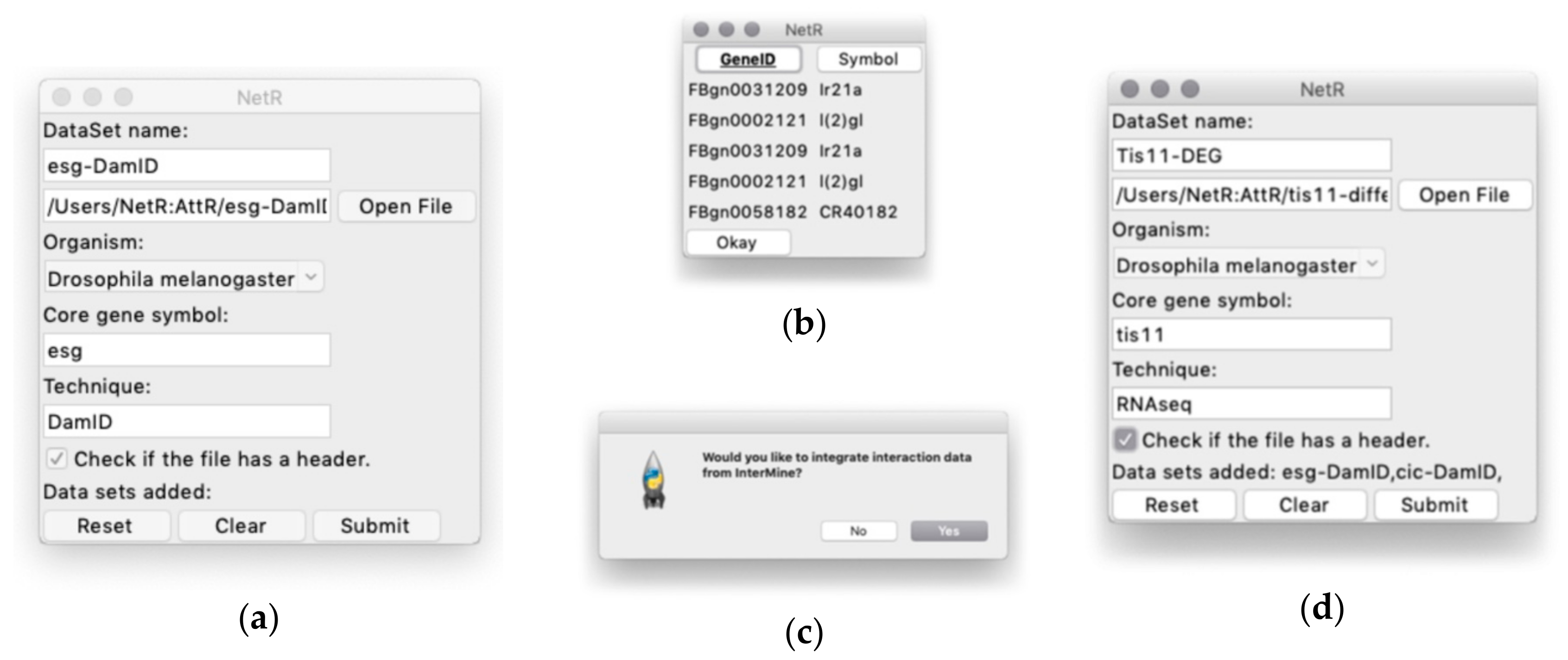
To facilitate the use of these valuable but less accessible data, we have developed two open source and easy-to-use computer programs (NetR and AttR) that allow users to combine diverse lists of genes or proteins into a network file that can later be mapped and visualized as a network of hypothetical interactions with Cytoscape [
6].
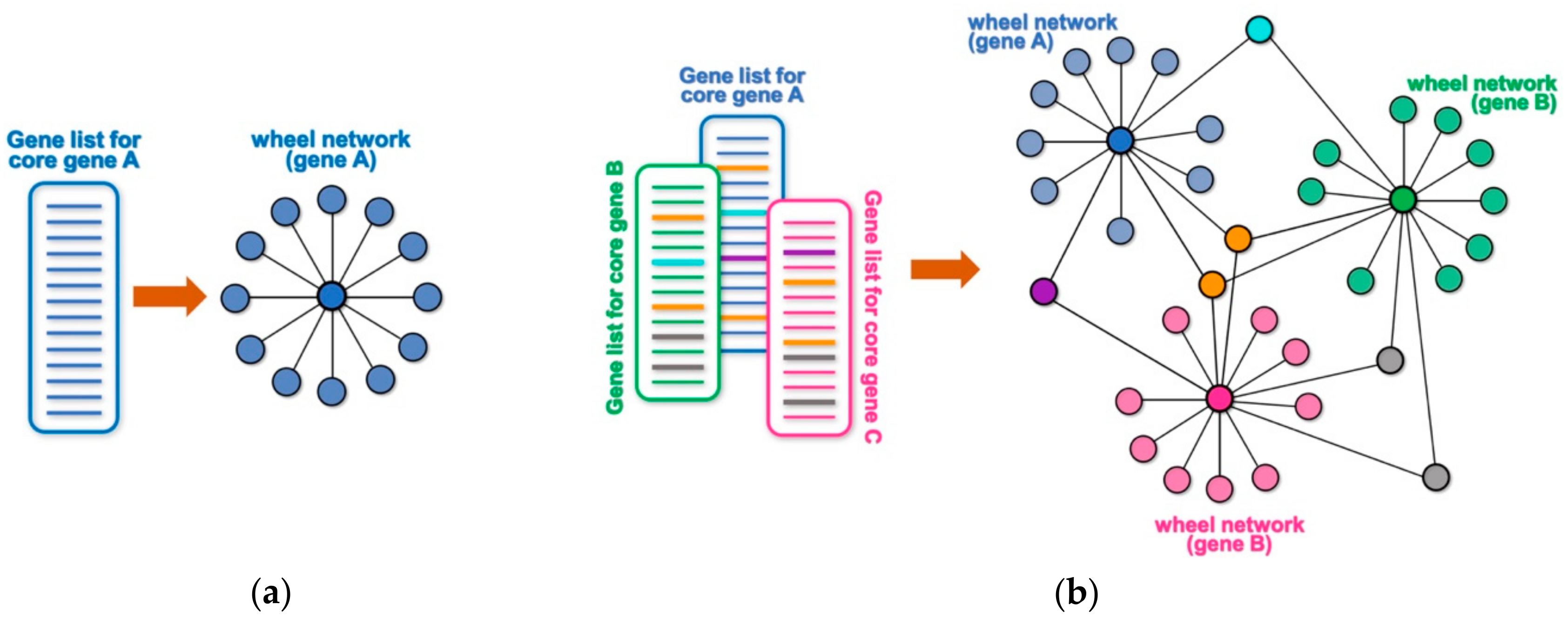
Many high throughput technologies are used to identify lists of genes or proteins that interact with or are somewhat targeted by a core gene (or protein) of interest. Examples may include genes that are differentially expressed following the experimental manipulation of the core gene (e.g., by overexpression, knock-down, mutation, etc.), the identification of putative transcriptional targets based on DNA-binding profile of a core gene (e.g., by chromatin immunoprecipitation and sequencing (ChIP-seq) or binding site search within promoter/enhancer regions), the identification of protein interactors of a protein of interest by a large-scale proteomic screen (e.g., immunoprecipitation followed by mass spectrometry analysis, yeast two-hybrid screens, etc.), the identification of genetic interactors by a genetic screen, or a screen for putative targets of an enzyme (e.g., by phosphoproteome analysis, peptide sequence search, etc.). Any of these unidimensional lists of targets or interactors for a core gene or protein can be conceptualized as a wheel network, where the core gene is at the center of the wheel and the rim of the wheel represents its corresponding targets or interactors (
Figure 1a). For example, a ChIP-seq experiment for a core transcription factor will generate a list of its putative target genes based on their proximity to the identified DNA-binding peaks. The obtained list of putative targets can be represented as a wheel network with the transcription factor at its hub and all the putative targets at the rim of the wheel. Likewise, knocking down the expression of a given cytoplasmic kinase by RNA interference, followed by transcriptome profiling through RNA-sequencing (RNA-seq) will generate a wheel network with the targeted kinase at its hub and all the differentially expressed genes at the rim. When multiple lists are combined, any targets that are shared among different wheel networks will become nodes that connect them into a larger network (
Figure 1b and
Figure S1). Such an approach can allow for a simpler and quicker visual identification of a smaller set of genes with hypothetically key regulatory roles, and NetR allows users to automatically combine any number of unidimensional lists into integrated network files that map all of the interactions between nodes in the combined network. The output produced by NetR is a comma-separated values (CSV) table which is easily readable by the open-source network visualization program Cytoscape [
6].
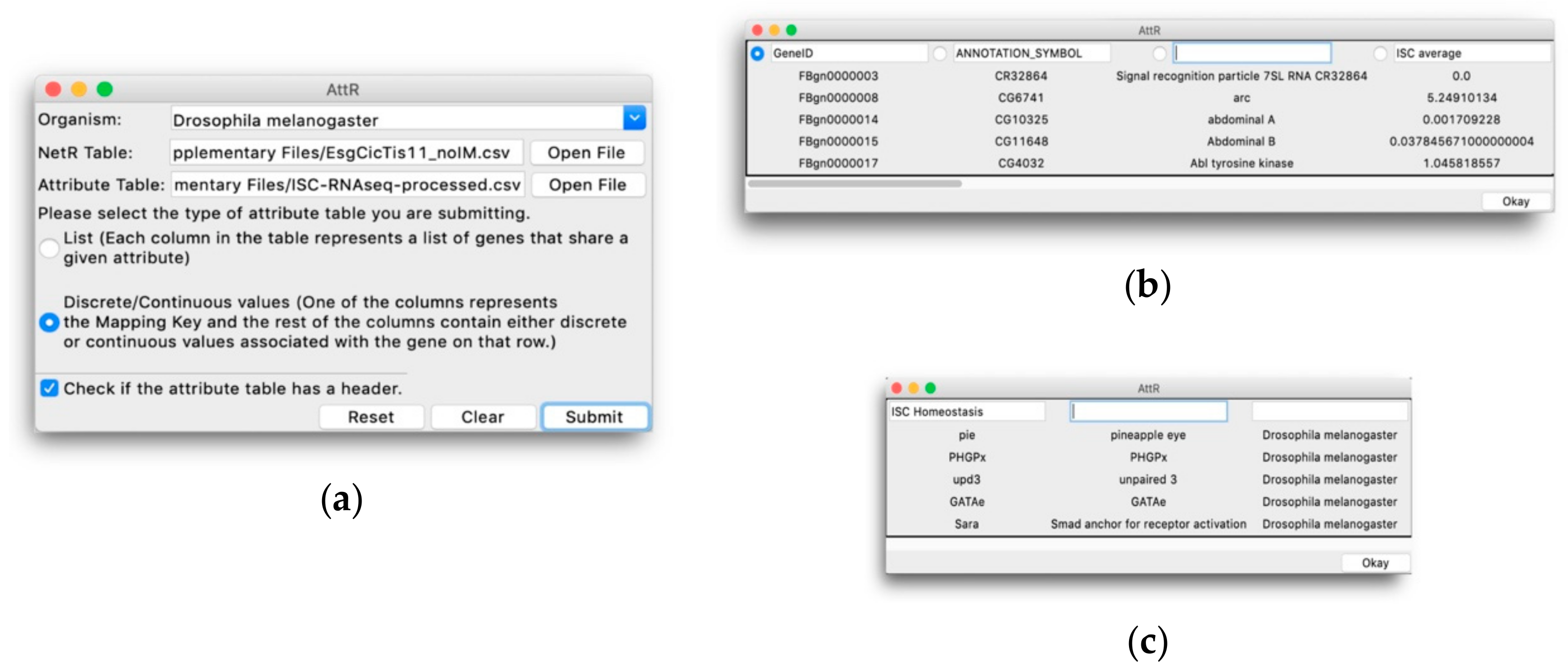
In addition to visualizing the connections between various nodes in a network, Cytoscape also allows users to import node attributes, such as membership to a gene/protein family, expression levels, cellular localization, etc. Once imported, these attributes can be used to identify nodes with specific attributes based on different colors, shapes, sizes, etc. AttR allows users to generate an all-in-one attribute table containing all the attributes for nodes in a combined NetR network, with up-to-date gene identifiers and properly formatted to be used with Cytoscape. Here we share and demonstrate how to use NetR and AttR, and how their use enabled us to integrate valuable data that were publicly available but inaccessible to other bioinformatics tools. We also show how their use allowed us to identify a subset of genes statistically enriched for biological features that make them interesting candidates for further studies, and that would have likely remained unidentified using classical approaches to data metanalysis.
4. Discussion
Recent technological advances have allowed researchers to routinely produce large amounts of genome-scale data from a wide range of experimental systems. While several tools exist for querying, retrieving, analyzing and sometimes integrating public datasets, the generation of system-wide data generally outpaces its re-utilization in subsequent studies, most notably among the same experimental biologists who generated the data in the first place. Such data underutilization may in part result from the lack of consistency in the way that datasets are tagged, stored and annotated [
15]. The latter may be particularly true about processed datasets that are available through publication, which are often hard to find through literature searches and are not easily analyzable through standard bioinformatics approaches due to data formatting inconsistencies. Furthermore, even when researchers are aware of several genome-scale studies by colleagues in their field, they may still be discouraged from more carefully re-analyzing them if they lack the programming skills that are often required to reformat and process public datasets.
Here we introduce two free and easy-to-use computer programs (NetR and AttR) that allow users to combine diverse lists of genes or proteins into networks that can be mapped with Cytoscape [
6]. While a certain degree of data processing was still required to use NetR and AttR, they involved simple downloading, copy/pasting and sorting operations with familiar computer programs and internet browsers, all of which are much better aligned with the computer skills of most experimental biologists.
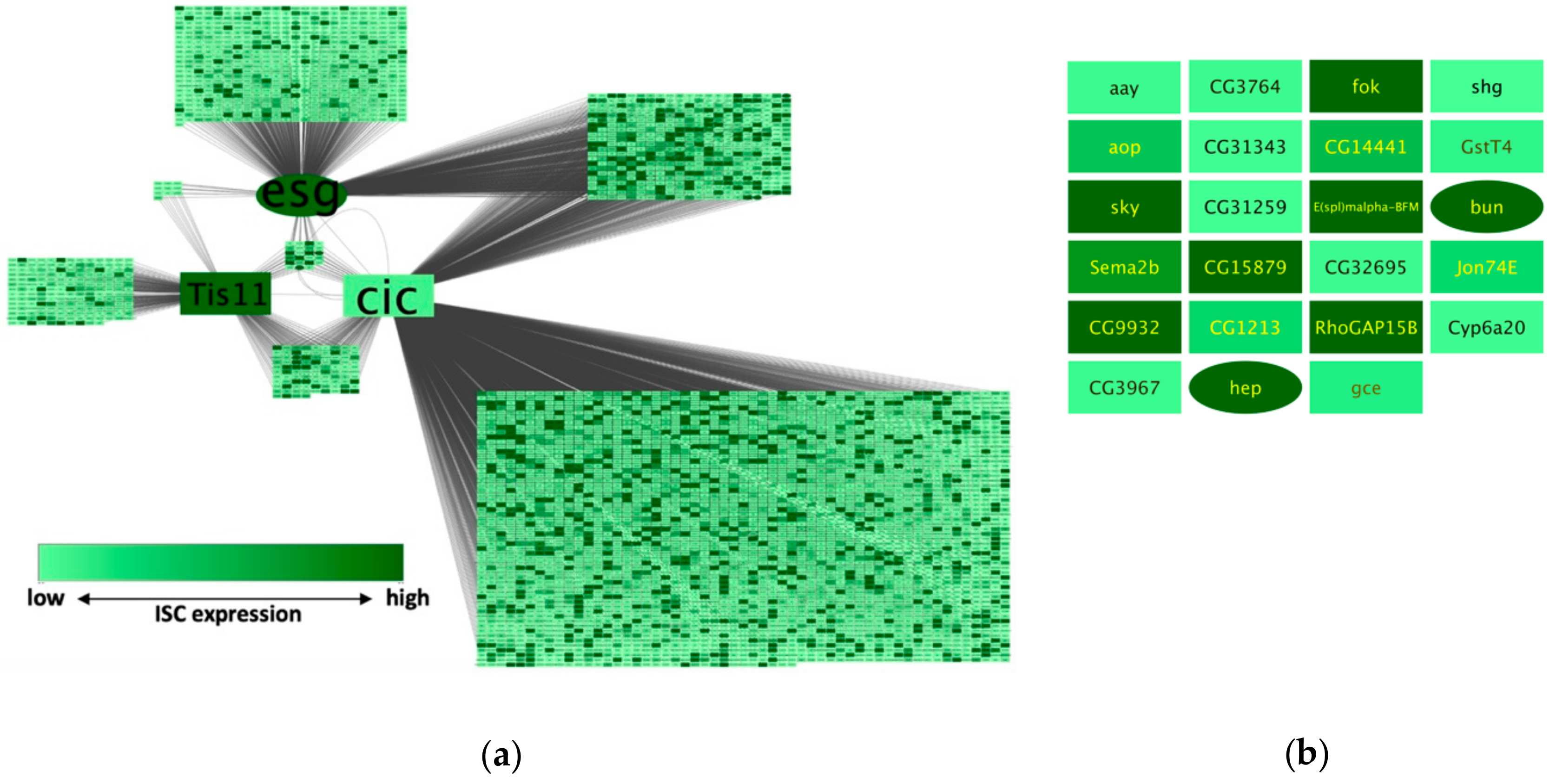
To demonstrate the features and applicability of NetR and AttR, we combined three independent datasets related to the genetic regulation of intestinal stem cells in the model organism
Drosophila melanogaster [
7,
8,
9]. Our approach allowed us to identify a small subset of targets that are putatively co-regulated by all three of the chosen master regulators in these cells (
Figure 4b), which makes them interesting candidates for validation and further experimental work. Interestingly, there was almost no overlap between the identified set of putatively co-regulated targets and a similar set of candidates that would have been chosen from each original dataset based on more traditional data mining approaches (e.g., significance ranking). When each original dataset was ranked based on their corresponding signal intensity (peak intensity ratio for Esg and Cic targets, and a ratio of fold-change in expression for Tis11), and the 23 top targets from each set were compared to the 23 putatively co-regulated targets identified by NetR, there was only one target (bun) shared between the Cic and NetR sets. Therefore, integrating different datasets allowed us to identify a vastly novel set of interesting candidates for further studies. Furthermore, the subset identified with NetR was significantly enriched for genes highly expressed in intestinal cells, as determined by an unrelated study [
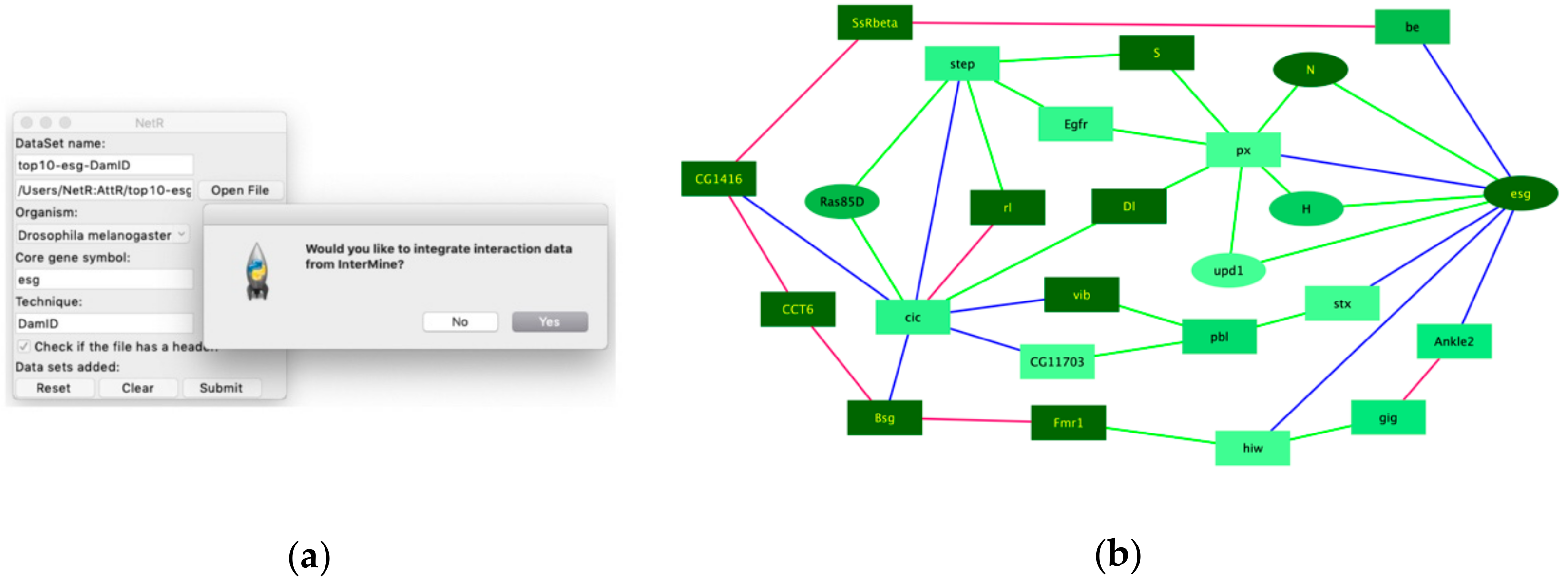
10], as well as genes curated under the “intestinal stem cell homeostasis” by the Gene Ontology Consortium. Of course, out data await both validation (i.e., to independently confirm that the putatively co-regulated targets are indeed regulated by each of the master regulators) and further functional studies (i.e., to determine whether they play a functional role in regulating intestinal stem cells in the fly intestine). However, if we consider expression as a proxy for the likelihood that a candidate might play a regulatory role in a cell of interest, then our findings would indicate that our intersection of independent but related datasets enhanced the discovery rate of biologically significant targets relative to the individual studies that contributed to the intersection. Similarly, integrating InterMine data when creating a NetR network allowed us to identify a set of 25 highly interconnected genes (
Figure 5b), which were also significantly enriched for genes classified under “Intestinal stem cell homeostasis” by Gene Ontology. Interestingly, 15 of these 25 genes were not even present in the original datasets, which represents an even clearer example of exploiting existing public data for identifying priority candidates for further studies. Our observations above, while interesting and encouraging, should be considered as anecdotal. We have not repeated our analyses with enough additional datasets to more confidently determine how often NetR/AttR lead to the identification of pursuable candidates that are both novel (i.e., that were not identified in the original datasets via traditional methods) and enriched for a biologically relevant feature. In this regard, it is important to emphasize that,
sensu stricto, neither NetR nor AttR represent discovery tools (i.e., neither program generates new knowledge based on available data). Instead, we prefer to think of NetR and AttR as purely heuristic tools, i.e., tools that facilitate the formulation of testable hypotheses based on existing data. For instance, when we state that NetR allowed for the identification of putatively co-regulated targets of Esg, Cic and Tis11, we are inherently reckoning that they still require experimental validation.
Of course, other tools exist for the mapping, visualizing and analyzing genetic or protein networks, many of which also offer users the opportunity to integrate interaction data from a series of existing sources and biological species. For instance, the Agilent application within Cytoscape allows users to submit a list of molecular entities that are used by multiple scientific literature search engines to retrieve documents in which the query terms are associated to other entities through specific association terms of interest. The associations detected by the Agilent app are then collected into a Cytoscape network, in which the sentences used to support each association are stored as edge attributes [
16]. OmicsNet (
https://www.omicsnet.ca/) allows users to upload lists of molecular identifiers and map retrieved molecular interactions of 4 different types (protein-protein, transcription factor-gene, miRNA-target gene and metabolite-protein) [
17]. In EsyN (
http://www.esyn.org/), users can upload a list of genes or proteins from diverse species and generate network models based on genetic and protein interaction data retrieved from InterMine and BioGrid (
https://thebiogrid.org/)—i.e., using a similar approach to integrating InterMine data when uploading gene lists to NetR [
18]. Lastly, GeneMania (
https://genemania.org/) (which can also be installed as an optional App in Cytoscape) offers a very powerful and user-friendly interface to find genes that are related to a list of genes provided by the user, based on association data that include protein and genetic interactions, curated pathways, co-expression and co-localization data [
19]. Each of these, and additional resources, represent different approaches to mining publicly available data based on diverse association criteria. We propose that NetR/AttR complement these powerful tools in three main ways.
They offer an opportunity to integrate publicly available or unpublished data that exist in formats that are less amenable to database cross-referencing or querying by literature search engines.
In the process of preparing datasets for uploading into NetR/AttR, users can exercise some degree of control over what nodes will be integrated into the network (e.g., by deciding on cutoffs of significance or selection criteria)
Users can use their expertise and judgement to integrate datasets that cross-referencing algorithms may miss, based on dataset annotation.
In this regard, NetR/AttR should be considered as complementary to other biological data management and processing tools, providing a somewhat higher degree of control over the data being integrated into a network model (although it should be emphasized that some of the resources mentioned above offer post-mapping network editing features that allow users to discard nodes or edges based on filtering criteria).
Each of the tools mentioned above presents strengths and limitations. Perhaps more interestingly, they all make use of different datasets, repositories and retrieval protocols, which gives rise to non-overlapping network models. For instance, when
esg,
cic and
Tis11 were used as lists of query genes for Agilent, EsyN and GeneMania, the network models retrieved were not highly similar to each other, nor to the one that we generated using NetR/AttR. Most notably, the Agilent/Cytoscape, EsyN and GeneMania networks did not include the experimental data generated by our source studies [
7,
8,
9], which are publicly available but not accessible to their cross-referencing algorithms. Thus, we believe that NetR and AttR can fill a significant gap in our ability to generate new testable hypotheses based on published observations, which could be deemed as important to scientific progress as the generation of new data per se.
One perceived limitation of NetR and AttR is that both are separate programs that simply produce network and attribute files to be used with Cytoscape. It is therefore reasonable to imagine that both programs would be more useful if directly integrated into Cytoscape as applications. However, we believe that Cytoscape integration would limit the flexibility and applicability of the programs, both of which were conceived and created to be open-source and easily customizable to serve additional purposes. While the NetR and AttR outputs work well with Cytoscape by design, the data in them could be useful outside of Cytoscape, particularly for more advanced users that may want to integrate them into their own bioinformatics pipelines. Furthermore, more advanced users may want to consider introducing the REST API into the NetR and AttR code, in order to call on powerful Cytoscape functions to work on the NetR and AttR files as part of their independent bioinformatic pipelines. In summary, we hope that more advanced users in the research and bioinformatics community feel compelled to use, experiment with, and improve NetR and AttR, in any number of unexpected and creative ways, including but not limited to full Cytoscape integration.
Ten years ago, while reflecting on a minimal bioinformatics skill set that modern biologists should possess, Tan et al. wondered how undergraduate and graduate life science students, already charged with having to learn the fundamentals and latest developments in various subdisciplines, could find the time to cope with additional bioinformatic and programming demands [
20]. These challenges are even more true nowadays, especially when the ease and speed with which new data are generated make it harder for students to keep up with the ever-expanding developments in modern biology. The constant advances in “-omics” techniques also make it increasingly challenging, even for trained bioinformaticians, to keep up with the diverse data structures and mathematical frameworks in modern bioinformatics. Perhaps even more worryingly, it is becoming harder to fill the widening gap between bioinformatics and hypothesis-driven experimental research. We acknowledge that NetR/AttR represent an overly simplistic approach to deal with heterogeneous datasets. However, and as indicated above, we developed NetR and AttR as heuristic tools for the generation of testable hypotheses based on largely unexploited datasets. In other words, NetR and AttR should not be used to answer biological questions, but to ask new ones; and we now wish to share these tools with experimental biologists at large, with the expectation that our programs will facilitate significant advances in their research efforts.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




