De Novo Sequencing, Assembly, and Annotation of Four Threespine Stickleback Genomes Based on Microfluidic Partitioned DNA Libraries

 , and
, and
Abstract
:1. Introduction
2. Methods and Materials
2.1. Stickleback Samples, DNA Library Preparation, Sequencing
2.2. Genome Assembly and Annotation
2.3. Comparative Sequence Alignment
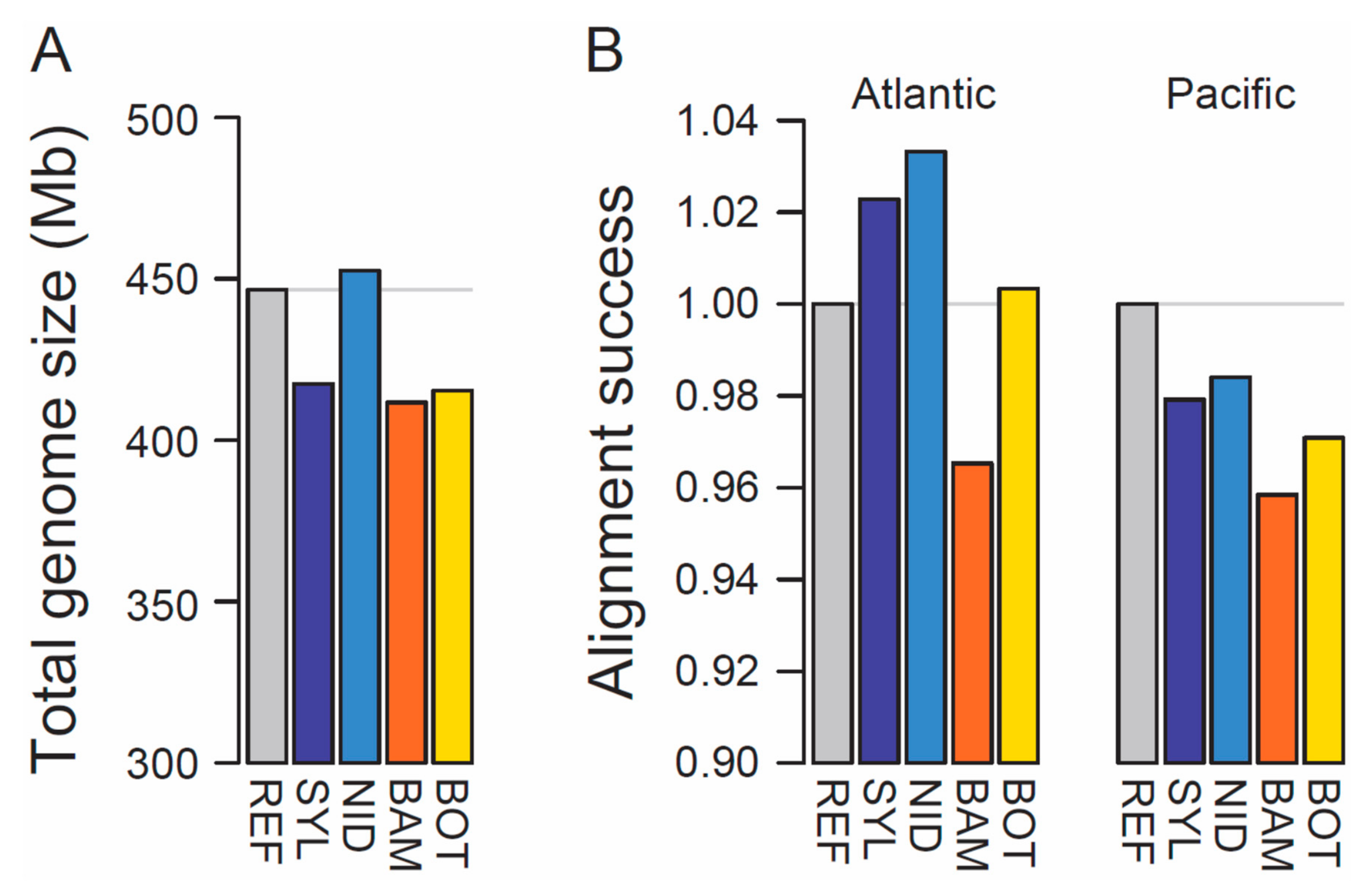
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bell, M.A.; Foster, S.A. The Evolutionary Biology of the Threespine Stickleback; Oxford University: Oxford, UK, 1994. [Google Scholar]
- Biology of the Three-Spined Stickleback; Östlund-Nilsson, S.; Mayer, I.; Huntingford, F.A. (Eds.) CRC: Boca Raton, FL, USA, 2007. [Google Scholar]
- McKinnon, J.S.; Rundle, H.D. Speciation in nature: The threespine stickleback model systems. Trends Ecol. Evol. 2002, 17, 480–488. [Google Scholar] [CrossRef]
- Peichel, C.L. Fishing for the secrets of vertebrate evolution in threespine sticklebacks. Dev. Dyn. 2005, 234, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Cresko, W.A.; McGuigan, K.L.; Phillips, P.C.; Postlethwait, J.H. Studies of threespine stickleback developmental evolution: Progress and promise. Genetica 2007, 129, 105–126. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S.; et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesti, M.; Moser, D.; Berner, D. Recombination in the threespine stickleback genome - patterns and consequences. Mol. Ecol. 2013, 22, 3014–3027. [Google Scholar] [CrossRef] [PubMed]
- Glazer, A.M.; Killingbeck, E.E.; Mitros, T.; Rokhsar, D.S.; Miller, C.T. Genome assembly improvement and mapping convergently evolved skeletal traits in sticklebacks with genotyping-by-sequencing. G3 2015, 5, 1463–1472. [Google Scholar] [CrossRef]
- Peichel, C.L.; Sullivan, S.T.; Liachko, I.; White, M.A. Improvement of the threespine stickleback genome using a Hi-C-based proximity-guided assembly. J. Hered. 2017, 108, 693–700. [Google Scholar] [CrossRef]
- Peichel, C.L.; Ross, J.A.; Matson, C.K.; Dickson, M.; Grimwood, J.; Schmutz, J.; Myers, R.M.; Mori, S.; Schluter, D.; Kingsley, D.M. The master sex-determination locus in threespine sticklebacks is on a nascent Y chromosome. Curr. Biol. 2004, 14, 1416–1424. [Google Scholar] [CrossRef]
- Jones, S.J.M.; Taylor, G.A.; Chan, S.; Warren, R.L.; Hammond, S.A.; Bilobram, S.; Mordecai, G.; Suttle, C.A.; Miller, K.M.; Schulze, A.; et al. The genome of the beluga whale (Delphinapterus leucas). Genes 2017, 8, 378. [Google Scholar] [CrossRef]
- Berner, D.; Roesti, M.; Hendry, A.P.; Salzburger, W. Constraints on speciation suggested by comparing lake-stream stickleback divergence across two continents. Mol. Ecol. 2010, 19, 4963–4978. [Google Scholar] [CrossRef]
- Higham, T.E.; Jamniczky, H.A.; Jagnandan, K.; Smith, S.J.; Barry, T.N.; Rogers, S.M. Comparative dynamics of suction feeding in marine and freshwater three-spined stickleback, Gasterosteus aculeatus: Kinematics and geometric morphometrics. Biol. J. Linn. Soc. 2017, 122, 400–410. [Google Scholar] [CrossRef]
- Hendry, A.P.; Taylor, E.B. How much of the variation in adaptive divergence can be explained by gene flow? An evaluation using lake-stream stickleback pairs. Evolution 2004, 58, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.; Coombe, L.; Warren, R.L.; Chu, J.; Birol, I. ARCS: Scaffolding genome drafts with linked reads. Bioinformatics 2017, 34, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Tzvetkova, A.; Morgenstern, B. AUGUSTUS at EGASP: Using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006, 7, S11. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinformatics 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simao, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Berner, D.; Ammann, M.; Spencer, E.; Rüegg, A.; Lüscher, D.; Moser, D. Sexual isolation promotes divergence between parapatric lake and stream stickleback. J. Evol. Biol. 2017, 30, 401–411. [Google Scholar] [CrossRef]
- Lavin, P.A.; McPhail, J.D. Parapatric lake and stream sticklebacks on northern Vancouver Island: Disjunct distribution or parallel evolution? Can. J. Zool. 1993, 71, 11–17. [Google Scholar] [CrossRef]
- Hendry, A.P.; Taylor, E.B.; McPhail, J.D. Adaptive divergence and the balance between selection and gene flow: Lake and stream stickleback in the Misty system. Evolution 2002, 56, 1199–1216. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Assembly | SYL | NID | BAM | BOT |
|---|---|---|---|---|
| Region | Atlantic | Atlantic | Pacific | Pacific |
| Habitat type | Marine | Freshwater | Marine | Freshwater |
| Locality [Reference] | List, Sylt, Germany | Aach stream, Switzerland [12] | Bamfield Inlet, Vancouver Island, Canada [13] | Boot Lake, Vancouver Island, Canada [14] |
| Geographic coordinates | 55°01′49.04″ N, 8°25′37″ E | 47°33′29.25″ N, 9°16′42.38″ E | 48°49′12.69″ N, 125°8′57.9″ W | 50°03′00.2″ N, 125°32′27.4″ W |
| Number of scaffolds | 15,853 | 10,246 | 25,430 | 18,433 |
| N50 (Mb) | 0.396 | 3.636 | 0.446 | 0.307 |
| Longest scaffold (Mb) | 3.12 | 16.02 | 4.33 | 3.81 |
| Total assembly length (Mb) (gapped length in parentheses) | 417.5 (431.8) | 452.5 (467.5) | 411.7 (445.7) | 414.9 (427.3) |
| Number of annotated genes with experimental evidence | 18,513 | 19,928 | 17,789 | 18,413 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berner, D.; Roesti, M.; Bilobram, S.; Chan, S.K.; Kirk, H.; Pandoh, P.; Taylor, G.A.; Zhao, Y.; Jones, S.J.M.; DeFaveri, J. De Novo Sequencing, Assembly, and Annotation of Four Threespine Stickleback Genomes Based on Microfluidic Partitioned DNA Libraries. Genes 2019, 10, 426. https://doi.org/10.3390/genes10060426
Berner D, Roesti M, Bilobram S, Chan SK, Kirk H, Pandoh P, Taylor GA, Zhao Y, Jones SJM, DeFaveri J. De Novo Sequencing, Assembly, and Annotation of Four Threespine Stickleback Genomes Based on Microfluidic Partitioned DNA Libraries. Genes. 2019; 10(6):426. https://doi.org/10.3390/genes10060426
Chicago/Turabian StyleBerner, Daniel, Marius Roesti, Steven Bilobram, Simon K. Chan, Heather Kirk, Pawan Pandoh, Gregory A. Taylor, Yongjun Zhao, Steven J. M. Jones, and Jacquelin DeFaveri. 2019. "De Novo Sequencing, Assembly, and Annotation of Four Threespine Stickleback Genomes Based on Microfluidic Partitioned DNA Libraries" Genes 10, no. 6: 426. https://doi.org/10.3390/genes10060426
APA StyleBerner, D., Roesti, M., Bilobram, S., Chan, S. K., Kirk, H., Pandoh, P., Taylor, G. A., Zhao, Y., Jones, S. J. M., & DeFaveri, J. (2019). De Novo Sequencing, Assembly, and Annotation of Four Threespine Stickleback Genomes Based on Microfluidic Partitioned DNA Libraries. Genes, 10(6), 426. https://doi.org/10.3390/genes10060426