Global Profiling of lncRNAs Expression Responsive to Allopolyploidization in Cucumis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA Isolation, cDNA Library Preparation, and Illumina Sequencing
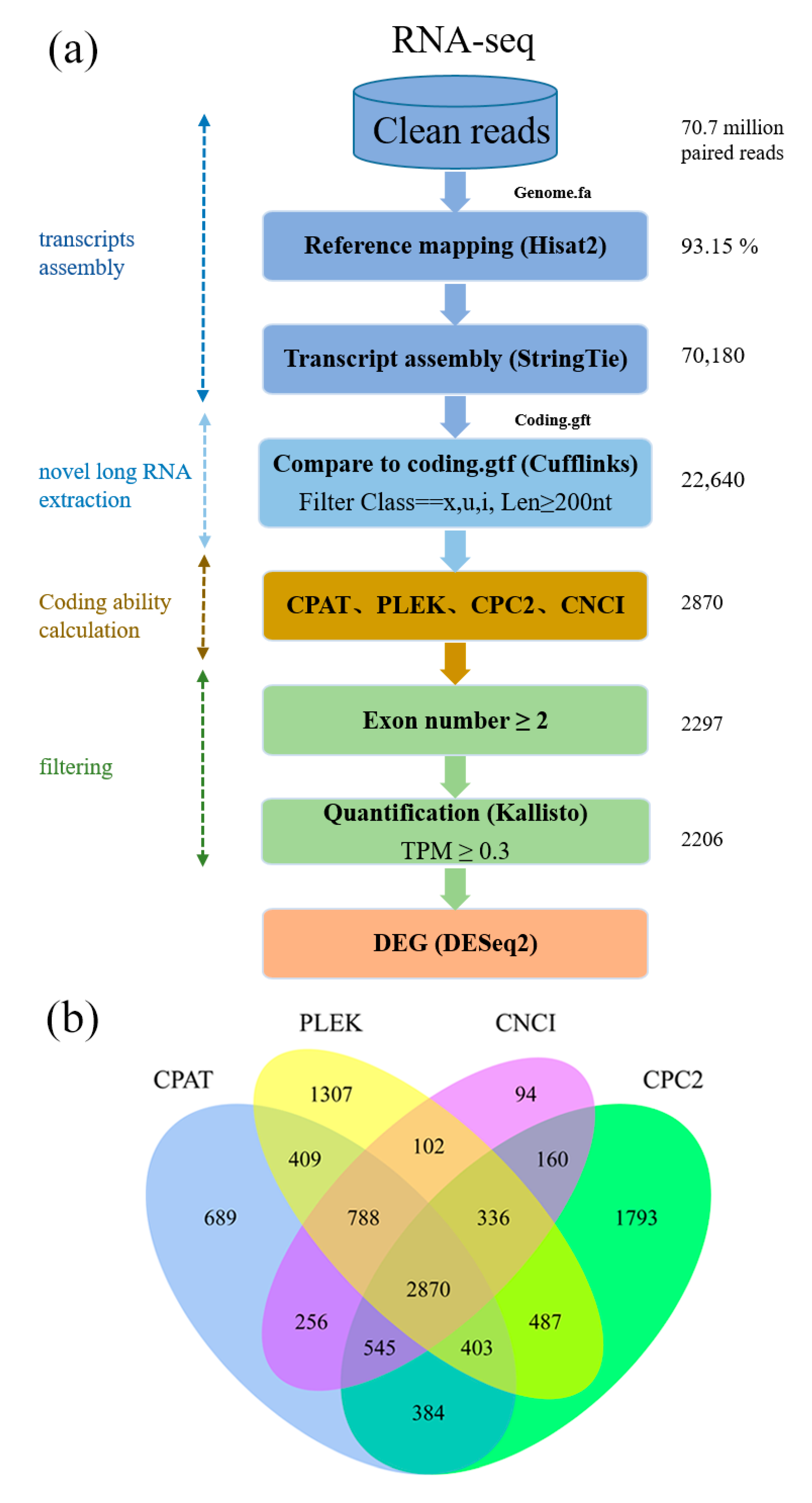
2.3. Transcriptome Assembling and Identification of lncRNAs
2.4. Differential Expression Analysis
2.5. Prediction of lncRNA Function
2.6. qRT-PCR Analysis for DE lncRNAs and Their Targets
2.7. Photosynthetic Parameters Measurements
3. Results
3.1. Identification of the Putative lncRNAs in C. hytivus Leaves
3.2. Sequence Characteristics of the lncRNAs
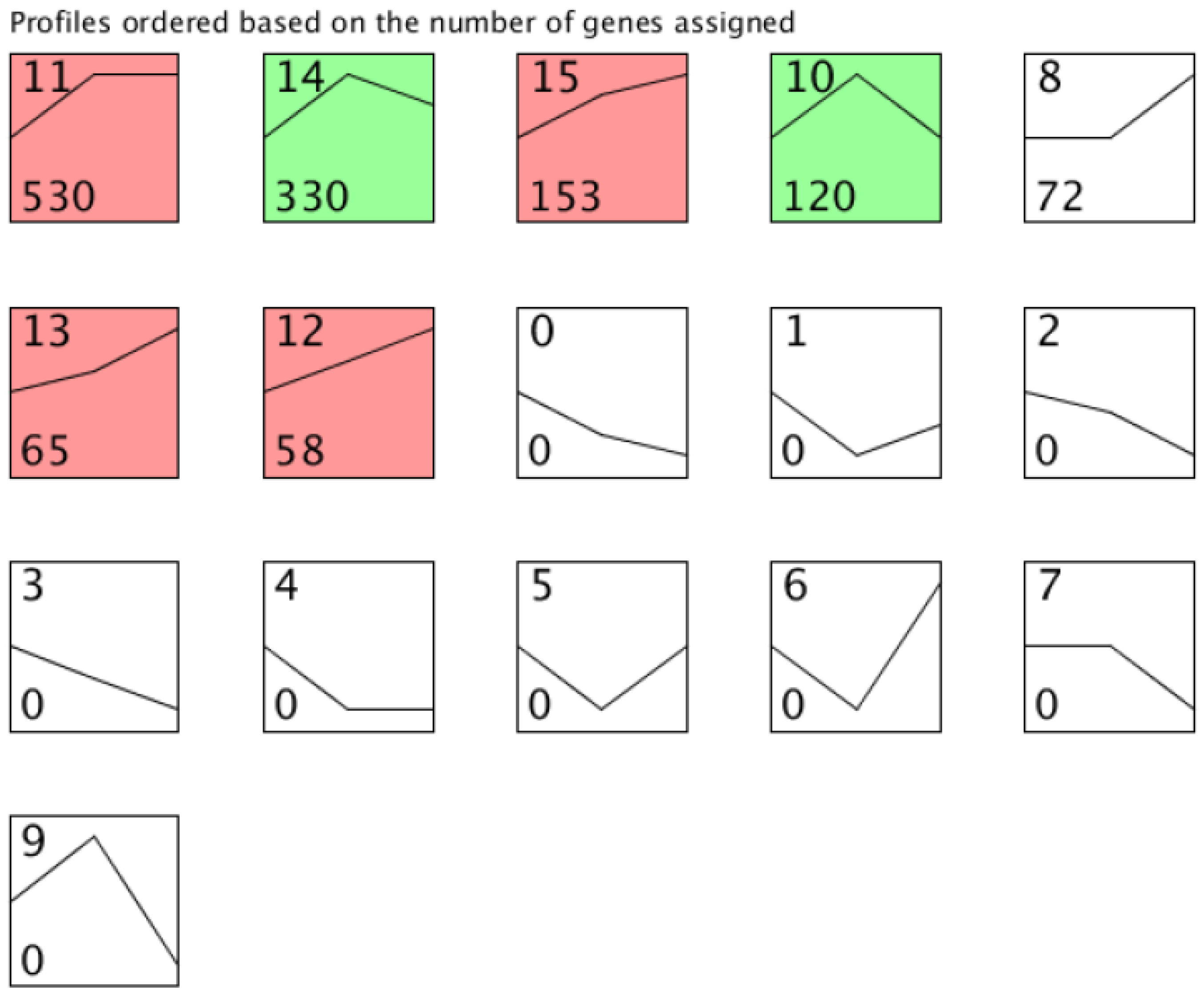
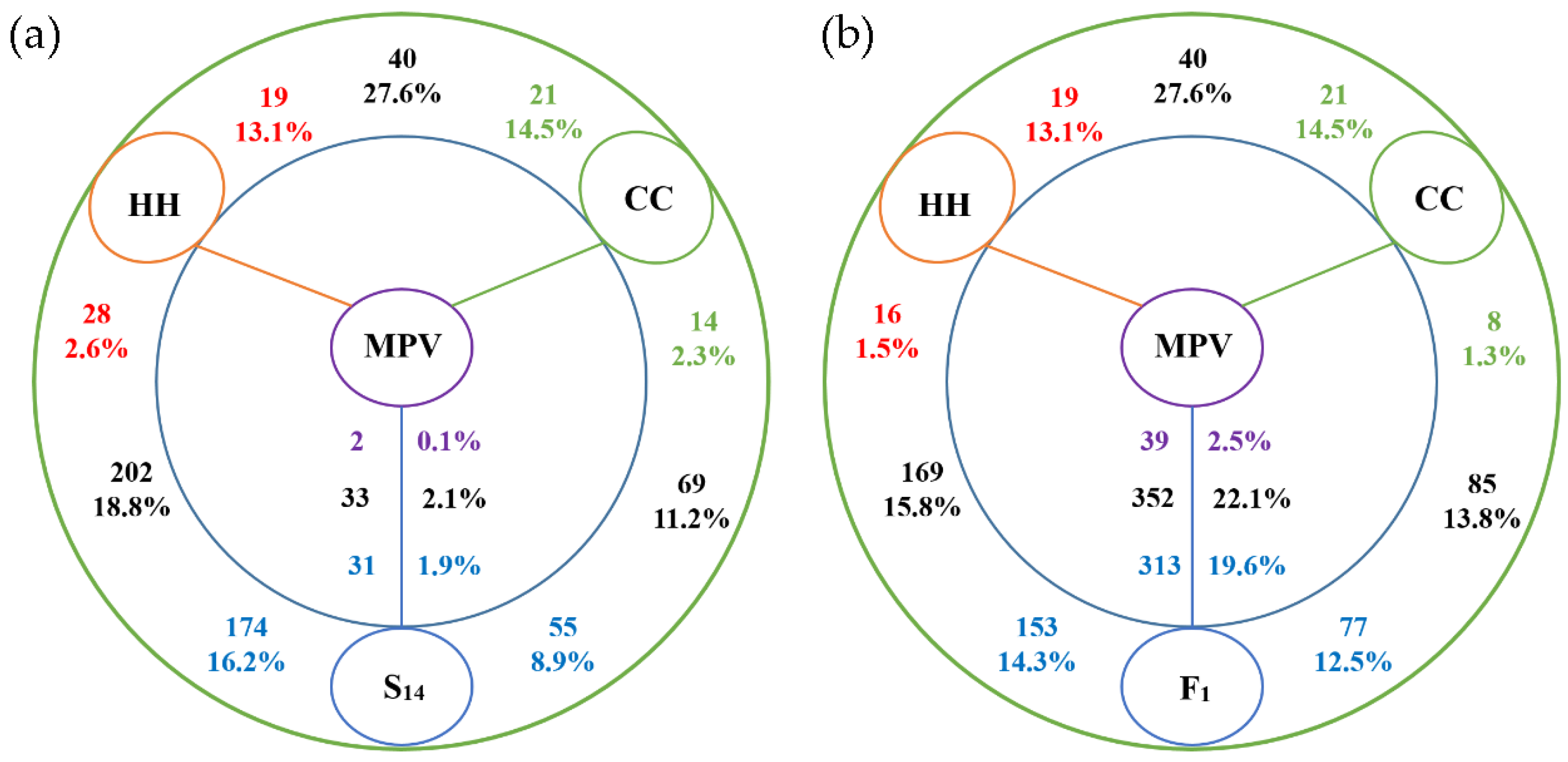
3.3. Profiling of lncRNA Expression in Interspecific Hybridization and Genome Duplication
3.4. Different Expression of lncRNA during Cucumis Allopolyploidization
3.5. Functional Analysis of Allopolyploidization-Related lncRNAs
3.6. qPCR Verification for DE-lncRNAs and Their Target Genes
4. Discussion
4.1. Genome-Wide Identification, Characterization of lncRNAs in Allotetraploid C. hytivus
4.2. Non-Additive lncRNA Expression under Intergeneric Hybridization and Polyploidization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.L.; Liu, D.C.; Wu, J.; Zhao, X.B.; Hao, M.; Geng, S.F.; Yan, J.; Jiang, X.X.; Zhang, L.Q.; Wu, J.Y.; et al. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 2014, 26, 1878–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Pauli, A.; Valen, E.; Lin, M.F.; Garber, M.; Vastenhouw, N.L.; Levin, J.Z.; Fan, L.; Sandelin, A.; Rinn, J.L.; Regev, A.; et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 2012, 22, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.L.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Necsulea, A.; Kaessmann, H. Evolutionary dynamics of coding and non-coding transcriptomes. Nat. Rev. Genet. 2014, 15, 734–748. [Google Scholar] [CrossRef]
- Chen, L.L.; Carmichae, G.G. Decoding the function of nuclear long noncoding RNAs. Curr. Opin. Cell Biol. 2010, 22, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Duret, L.; Chureau, C.; Samain, S.; Weissenbach, J.; Avner, P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science 2006, 312, 1653–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleutels, F.; Zwart, R.; Barlow, D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 2002, 415, 810–813. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segale, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Mach, J. The long-noncoding RNA ELENA1 functions in plant immunity. Plant Cell 2017, 29, 916. [Google Scholar] [CrossRef] [Green Version]
- Yatusevich, R.; Fedak, H.; Ciesielski, A.; Krzyczmonik, K.; Kulik, A.; Dobrowolska, G.; Swiezewski, S. Antisense transcription represses Arabidopsis seed dormancy QTL DOG1 to regulate drought tolerance. EMBO Rep. 2017, 18, 2186–2196. [Google Scholar] [CrossRef]
- Amor, B.B.; Wirth, S.; Merchan, F.; Laporte, P.; d’Aubenton-Carafa, Y.; Hirsch, J.; Maizel, A.; Mallory, A.C.; Lucas, A.; Deragon, J.M.; et al. Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009, 19, 57–69. [Google Scholar] [CrossRef] [Green Version]
- Matsui, A.; Ishida, J.; Morosawa, T.; Mochizuki, Y.; Kaminuma, E.; Endo, T.A.; Okamoto, M.; Nambara, E.; Nakajima, M.; Kawashima, M.; et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008, 49, 1135–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fan, X.; Lin, F.; He, G.; Terzaghi, W.; Zhu, D.; Deng, X.W. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc. Natl. Acad. Sci. USA 2014, 111, 10359–10364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; Garcia, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Ding, J.H.; Lu, Q.; Ouyang, Y.D.; Mao, H.L.; Zhang, P.B.; Yao, J.L.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Leitch, A.R.; Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef]
- Soltis, D.E.; Albert, V.A.; Leebens-Mack, J.; Bell, C.D.; Paterson, A.H.; Zheng, C.; Sankoff, D.; Depamphilis, C.W.; Wall, P.K.; Soltis, P.S. Polyploidy and angiosperm diversification. Am. J. Bot. 2009, 96, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, S.C.; Tyagi, A.P.; Thornton, G.M.; Pires, J.C.; Madlung, A. Allopolyploidization lays the foundation for evolution of distinct populations: Evidence from analysis of synthetic Arabidopsis allohexaploids. Genetics 2012, 191, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Chen, Z.J. Epigenetic and developmental regulation in plant polyploids. Curr. Opin. Plant Biol. 2015, 24, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, M.J.; Szadkowski, E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity (Edinb) 2013, 110, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Tao, X.Y.; Feng, S.L.; Wang, L.Y.; Hong, H.; Ma, W.; Shang, G.D.; Guo, S.S.; He, Y.X.; Zhou, B.L.; et al. LncRNAs in polyploid cotton interspecific hybrids are derived from transposon neofunctionalization. Genome Biol. 2018, 19, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Kirkbride, J.H. A new synthetic species of Cucumis (Cucurbitaceae) from interspecific hybridization and chromosome doubling. Brittonia 2000, 52, 315–319. [Google Scholar] [CrossRef]
- Chen, J.F.; Staub, J.E.; Tashiro, Y.; Isshiki, S.; Miyazaki, S. Successful interspecific hybridization between Cucumis sativus L. and C. hystrix Chakr. Euphytica 1997, 96, 413–419. [Google Scholar] [CrossRef]
- Chen, J.F.; Jeffrey, W.A.; Staub, J.E.; Skorupska, H.T.; Rhodes, B.B. A new synthetic amphidiploid in Cucumis from a C. sativus × C. hystrix F1 interspecific hybrid. Cucurbitaceae 1998, 1, 336–339. [Google Scholar]
- Sebastian, P.; Schaefer, H.; Telford, I.R.; Renner, S.S. Cucumber (Cucumis sativus) and melon (C. melo ) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc. Natl. Acad. Sci. USA 2010, 107, 14269–14273. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Chen, J.F. Changes of gene expression in early generations of the synthetic allotetraploid Cucumis × hytivus Chen et Kirkbride. Genet. Resour. Crop. Evol. 2009, 56, 1071–1076. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.P.; Gao, G. Cpc2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Zhang, J.Y.; Zhou, Z.Y. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.G.; Park, H.J.; Dasari, S.; Wang, S.Q.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.T.; Bu, D.C.; Zhao, G.G.; Yu, K.T.; Zhang, C.H.; Liu, Y.N.; Chen, R.S.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef]
- Shen, E.; Zhu, X.T.; Hua, S.J.; Chen, H.Y.; Ye, C.Y.; Zhou, L.H.; Liu, Q.; Zhu, Q.H.; Fan, L.J.; Chen, X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Krzywinski, M.; Schein, J.I. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Q.; Liu, C.N.; Yuan, X.Y.; Kang, S.L.; Miao, R.Y.; Xiao, H.; Zhao, G.G.; Luo, H.T.; Bu, D.H.; Zhao, H.T. Large-scale prediction of long noncoding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res. 2011, 39, 3864–3878. [Google Scholar] [CrossRef] [PubMed]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Wang, X.X.; Hyldgaard, B.; Zhu, Z.B.; Zhou, R.; Kjaer, K.H.; Ouzounis, T.; Lou, Q.F.; Li, J.; Cai, Q.S.; et al. Allopolyploidization in Cucumis contributes to delayed leaf maturation with repression of redundant homoeologous genes. Plant J. 2018, 94, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Q.; Hyldgaard, B.; Rosenqvist, E.; Ottosen, C.O.; Chen, J.F. Interspecific hybridization in Cucumis leads to the divergence of phenotypes in response to low light and extended photoperiods. Front. Plant. Sci. 2015, 6, 802. [Google Scholar] [CrossRef] [Green Version]
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [Green Version]
- Mayrose, I.; Zhan, S.H.; Rothfels, C.J.; Magnuson-Ford, K.; Barker, M.S.; Rieseberg, L.H.; Otto, S.P. Recently formed polyploid plants diversify at lower rates. Science 2011, 333, 1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Z.; Zhang, Z.T.; Jia, L.; Li, Z.A.; Li, J.; Lou, Q.F.; Chen, J.F. Molecular and cytogenetic analyses provide evidence of the introgression of chromosomal segments from the wild Cucumis hystrix into the cultivated cucumber through the bridge of a synthetic allotetraploid. Mol. Breed. 2017, 37, 89. [Google Scholar] [CrossRef]
- Zhang, K.J.; Wang, X.; Zhu, W.W.; Qin, X.D.; Xu, J.; Cheng, C.Y.; Lou, Q.F.; Li, J.; Chen, J.F. Complete resistance to powdery mildew and partial resistance to downy mildew in a Cucumis hystrix introgression line of cucumber were controlled by a co-localized locus. Theor. Appl. Genet. 2018, 131, 2229–2243. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Wang, X.; Liu, X.J.; Yang, S.Q.; Yu, X.Q.; Qian, C.T.; Li, J.; Lou, Q.F.; Chen, J.F. Candidate genes underlying the quantitative trait loci for root-knot nematode resistance in a Cucumis hystrix introgression line of cucumber based on population sequencing. J. Plant Res. 2019, 132, 813–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cheng, C.Y.; Li, Q.R.; Zhang, K.J.; Lou, Q.F.; Li, J.; Chen, J.F. Multi-omics analysis revealed that MAPK signaling and flavonoid metabolic pathway contributed to resistance against Meloidogyne incognita in the introgression line cucumber. J. Proteom. 2020, 220, 103675. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Zhuang, F.Y.; Liu, X.A.; Qian, C.T. Reciprocal differences of morphological and DNA characters in interspecific hybridization in Cucumis. Can. J. Bot. 2004, 82, 16–21. [Google Scholar] [CrossRef]
- Chen, L.Z.; Lou, Q.F.; Zhuang, Y.; Chen, J.F.; Zhang, X.Q.; Wolukau, J.N. Cytological diploidization and rapid genome changes of the newly synthesized allotetraploids Cucumis × hytivus. Planta 2007, 225, 603–614. [Google Scholar] [CrossRef]
- Jiang, B.; Lou, Q.F.; Chen, J.F. Genetic variation analysis of the newly synthesized allotetraploids Cucumis hytivus using retrotransposon-based SSAP markers. Acta Hortic. 2010, 871, 567–572. [Google Scholar] [CrossRef]
- Jiang, B.; Lou, Q.F.; Wu, Z.M.; Zhang, W.P.; Wang, D.; Mbira, K.G.; Weng, Y.Q.; Chen, J.F. Retrotransposon- and microsatellite sequence-associated genomic changes in early generations of a newly synthesized allotetraploid Cucumis. Plant. Mol. Biol. 2011, 77, 225–233. [Google Scholar] [CrossRef]
- Jiang, B.; Lou, Q.F.; Wang, D.; Wu, Z.M.; Zhang, W.P.; Chen, J.F. Allopolyploidization induced the activation of Ty1-copia retrotransposons in Cucumis hytivus, a newly formed Cucumis allotetraploid. Bot. Stud. 2011, 52, 145–152. [Google Scholar]
- Shen, J.; Kere, M.G.; Chen, J.F. Mitochondrial genome is paternally inherited in Cucumis allotetraploid (C. × hytivus) derived by interspecific hybridization. Sci. Hortic. 2013, 155, 39–42. [Google Scholar] [CrossRef]
- Zhuang, Y.; Chen, J.F.; Jahn, M. Expression and sequence variation of the cucumber Por gene in the synthesized allotetraploid Cucumis × hytivus. Mol. Biol. Rep. 2009, 36, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Z.; Chen, J.F. Changes of cytosine methylation induced by wide hybridization and allopolyploidy in Cucumis. Genome 2008, 51, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Z.; Zhao, Q.Z.; Qin, X.D.; Yang, S.Q.; Li, Z.A.; Li, J.; Lou, Q.F.; Chen, J.F. Identification of all homoeologous chromosomes of newly synthetic allotetraploid Cucumis × hytivus and its wild parent reveals stable subgenome structure. Chromosoma 2017, 126, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Zhu, Z.B.; Zhang, T.; Li, J.; Cheng, C.Y.; Lou, Q.F.; Ottosen, C.O.; Chen, J.F. High-throughput sequencing reveals the change of microRNA expression caused by allopolyploidization in Cucumis. Biol. Plant. 2020, 64, 104–109. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.D.; Sung, S. Long noncoding RNA: Unveiling hidden layer of gene regulatory networks. Trends Plant Sci. 2012, 17, 16–21. [Google Scholar] [CrossRef]
- Shumayla; Sharma, S.; Taneja, M.; Tyagi, S.; Singh, K.; Upadhyay, S.K. Survey of high throughput RNA-Seq data reveals potential roles for lncRNAs during development and stress response in bread wheat. Front. Plant Sci. 2017, 6, 1019. [Google Scholar]
- Chen, X.; Sun, S.; Liu, F.J.; Shen, E.H.; Liu, L.; Ye, C.Y.; Xiao, B.G.; Timko, M.; Zhu, Q.H.; Fan, L.J.; et al. A transcriptomic profile of topping responsive non-coding RNAs in tobacco roots (Nicotiana tabacum). BMC Genom. 2019, 20, 856. [Google Scholar] [CrossRef]
- Yao, S.L.; Liang, F.; Gill, R.A.; Huang, J.Y.; Cheng, X.H.; Liu, Y.Y.; Tong, C.B.; Liu, S.Y. A global survey of the transcriptome of the allopolyploid Brassica napus based on single molecule long-read isoform sequencing and Illumina-based RNA-seq data. Plant J. 2020, 103, 843–857. [Google Scholar] [CrossRef]
- Rošić, S.; Erhardt, S. No longer a nuisance: Long non-coding RNAs join CENP-A in epigenetic centromere regulation. Cell Mol. Life Sci. 2016, 73, 1387–1398. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.B.; Huang, W.; Xu, Y.C.; Zhou, Q.; Wang, S.H.; Ruan, J.; Huang, S.W.; Zhang, Z.H. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golicz, A.; Singh, M.B.; Bhalla, P.L. The long intergenic non-coding RNA (lincRNA) landscape of the soybean genome. Plant Physiol. 2017, 176, 01657. [Google Scholar]
- Vandivier, L.E.; Andeson, S.J.; Folev, S.W.; Gregory, B.D. The conservation and function of RNA secondary structure in plants. Annu. Rev. Plant. Biol. 2016, 67, 463–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.Z.; Yang, Y.F.; Li, R.; Fu, D.Q.; Wen, L.W.; Luo, Y.B.; Zhu, H.L. RNA sequencing and functional analysis implicate the regulatory role of long non-coding RNAs in tomato fruit ripening. J. Exp. Bot. 2015, 66, 4483–4495. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.L.; Du, Q.Z.; Chen, J.H.; Wang, Q.S.; Zhang, D.Q. Identification and allelic dissection uncover roles of lncRNAs in secondary growth of Populus tomentosa. DNA Res. 2017, 24, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Xiong, Y.Y.; Xue, Y.B.; Mao, M.Q.; Xiang, Y.X.; He, Y.H.; Rafique, F.; Hu, H.; Liu, J.W.; Li, X.; et al. Screening and characterization of long noncoding RNAs involved in the albinism of Ananas comosus var. bracteatus leaves. PLoS ONE 2019, 14, e0225602. [Google Scholar] [CrossRef]
- Henriques, R.; Wang, H.; Liu, J.; Boix, M.; Huang, L.F.; Chua, N.H. The antiphasic regulatory module comprising CDF5 and its antisense RNA FLORE links the circadian clock to photoperiodic flowering. N. Phytol. 2017, 216, 854–867. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Li, J.R.; Yang, Y.; Tan, C.; Zhu, Y.M.; Hu, L.; Qi, Y.J.; Lu, Z. Stress-responsive regulation of long noncoding RNAs polyadenylation in Oryza sativa. Plant J. 2017, 93, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Zhong, L.; Wu, X.M.; Fang, X.P.; Wang, J.B. Rapid alterations of gene expression and cytosine methylation in newly synthesized Brassica napus allopolyploids. Planta 2009, 229, 471–483. [Google Scholar] [CrossRef]
- Cheng, S.P.; Huang, Z.; Suo, Y.J.; Wang, J.; Kang, X.Y. Gene expression differences associated with growth vigor in Populus full-sib allotriploid progeny following manipulated first division restitution of the diploid maternal parent. Euphytica 2015, 203, 683–700. [Google Scholar] [CrossRef]
- Qi, X.Y.; Wang, H.B.; Song, A.P.; Jiang, J.F.; Chen, S.M.; Chen, F.D. Genomic and transcriptomic alterations following intergeneric hybridization and polyploidization in the Chrysanthemum nankingense × Tanacetum vulgare hybrid and allopolyploid (Asteraceae). Hortic. Res. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, A.K.; Tripathy, B.C. Temperature-stress-induced impairment of chlorophyll biosynthetic reactions in cucumber and wheat. Plant Physiol. 1998, 117, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertsson, P.Å. The structure and function of the chloroplast photosynthetic membrane—A model for the domain organization. Photosynth. Res. 1995, 46, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Hippler, M.; Klein, J.; Fink, A.; Allinger, T.; Hoerth, P. Towards functional proteomics of membrane protein complexes: Analysis of thylakoid membranes from Chlamydomonas reinhardtii. Plant J. 2010, 28, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Cheng, C.Z.; Lin, Y.L.; Xu, X.H.; Lai, Z.X. Genome-wide identification and characterization of mRNAs and lncRNAs involved in cold stress in the wild banana (Musa itinerans). PLoS ONE 2018, 13, e0200002. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.E.; Wegmann, B.; Wang, W.Y. Purification and characterization of glutamyl-tRNA synthetase: An enzyme involved in chlorophyll biosynthesis. Plant Physiol. 1990, 93, 1641–1649. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Yu, X.; Zhu, Z.; Zhai, Y.; Zhao, Q.; Meng, Y.; Li, J.; Lou, Q.; Chen, J. Global Profiling of lncRNAs Expression Responsive to Allopolyploidization in Cucumis. Genes 2020, 11, 1500. https://doi.org/10.3390/genes11121500
Wang P, Yu X, Zhu Z, Zhai Y, Zhao Q, Meng Y, Li J, Lou Q, Chen J. Global Profiling of lncRNAs Expression Responsive to Allopolyploidization in Cucumis. Genes. 2020; 11(12):1500. https://doi.org/10.3390/genes11121500
Chicago/Turabian StyleWang, Panqiao, Xiaqing Yu, Zaobing Zhu, Yufei Zhai, Qinzheng Zhao, Ya Meng, Ji Li, Qunfeng Lou, and Jinfeng Chen. 2020. "Global Profiling of lncRNAs Expression Responsive to Allopolyploidization in Cucumis" Genes 11, no. 12: 1500. https://doi.org/10.3390/genes11121500
APA StyleWang, P., Yu, X., Zhu, Z., Zhai, Y., Zhao, Q., Meng, Y., Li, J., Lou, Q., & Chen, J. (2020). Global Profiling of lncRNAs Expression Responsive to Allopolyploidization in Cucumis. Genes, 11(12), 1500. https://doi.org/10.3390/genes11121500