New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors

{kind=link}
{kind=link}
Abstract
:1. Neurofibromatosis Type 1 Syndrome and Associated Peripheral Nerve Sheath Tumors
2. Molecular Genetics of the NF1 Gene Product and MPNST
3. MPNST Model Systems
3.1. Genetically Engineered Mouse Models (GEMM)
3.2. Human MPNST Cell Lines and Patient Derived Xenografts
3.3. Human Cell-Based Models for MPNST
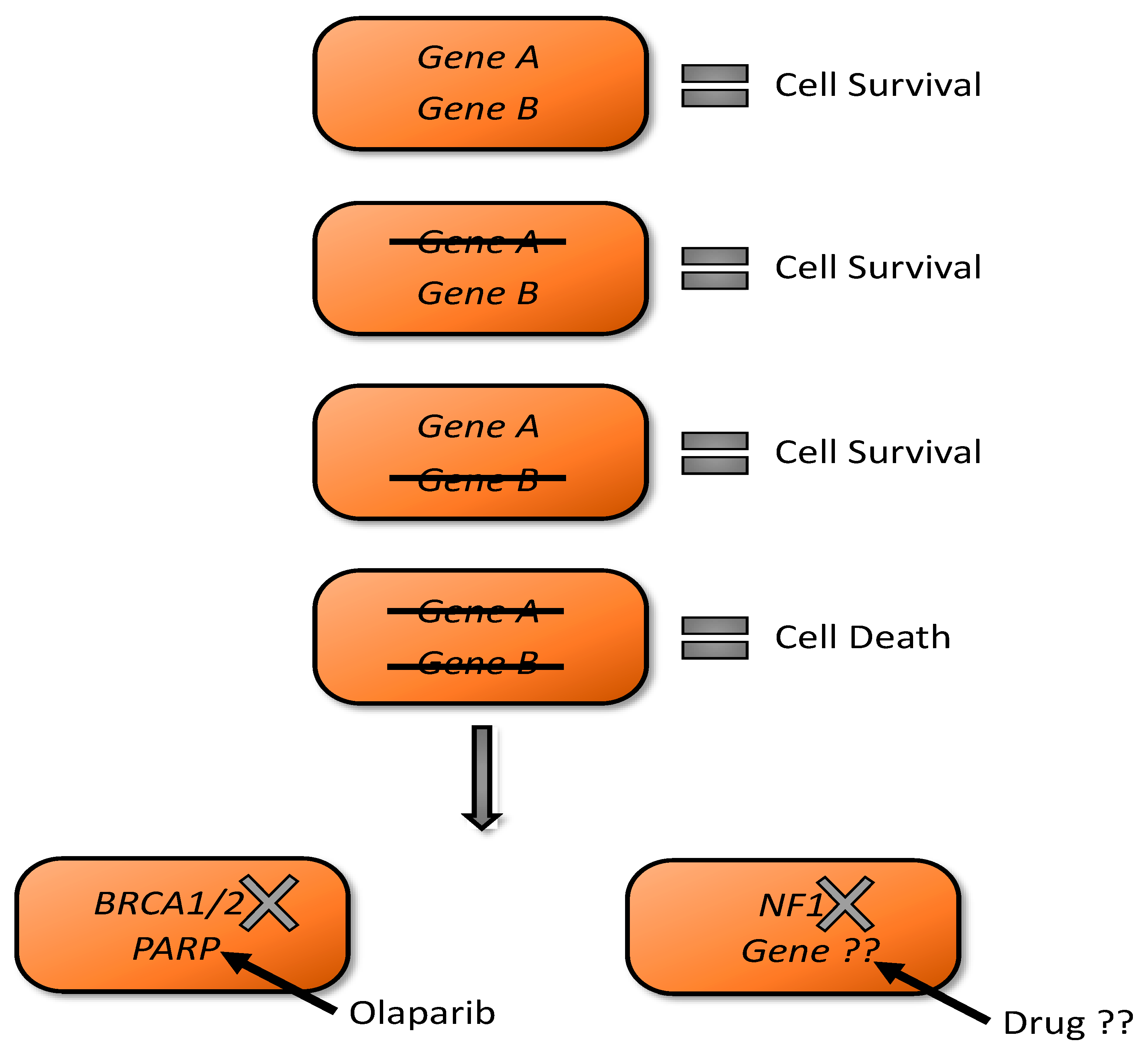
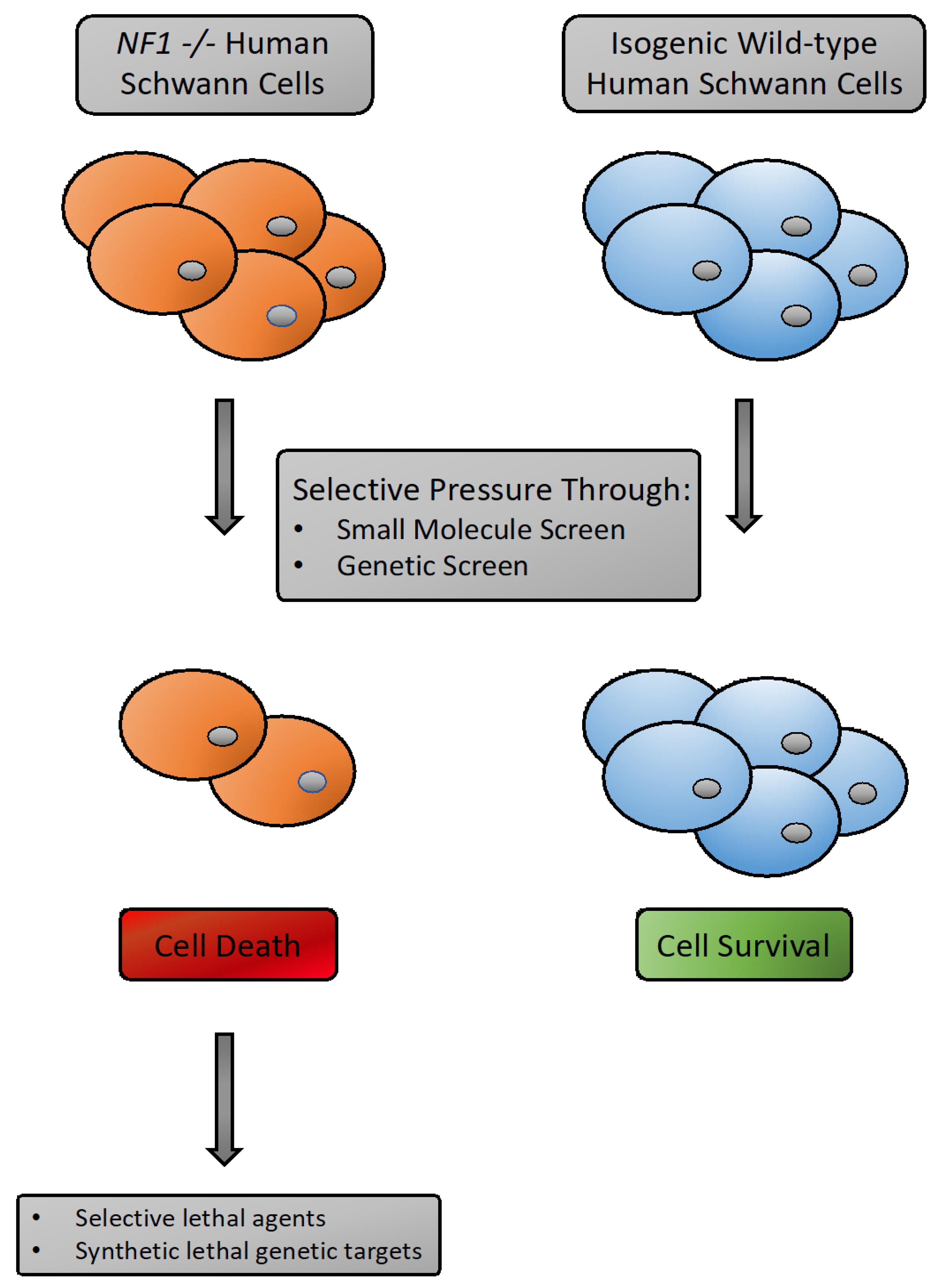
3.4. Synthetic Lethality as A Tool for NF1 Drug Discovery
3.5. Preclinical Development of New Therapies for NF1-Associated MPNST
3.6. Targeting Ras or Ras-Activated Signaling Pathways
3.7. Combination Signal Pathway Inhibition
3.8. Targeting Cyclin-Dependent Kinases for MPNST
3.9. Sensitivities Associated with Loss of PRC2 Function in MPNST Cells
3.10. Other Therapeutic Approaches
3.11. Comprehensive Pharmacological Profiling of Neurofibromatosis Type 1 Cancer Cell Lines
3.12. Synthetic Sensitivities Identified by Drug Screening in Nf1-Deficient Mouse Embryo Fibroblasts
4. Summary and Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis Primers. 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Martin, A.; Duat-Rodriguez, A. An Update on Neurofibromatosis Type 1: Not Just Cafe-au-Lait Spots and Freckling. Part II. Other Skin Manifestations Characteristic of NF1. NF1 and Cancer. Actas Dermosifiliogr. 2016, 107, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.F.; Weber, S.M.; Turner-Ivey, B.P.; Carroll, S.L. Recent Advances in the Diagnosis and Pathogenesis of Neurofibromatosis Type 1 (NF1)-associated Peripheral Nervous System Neoplasms. Adv. Anat. Pathol. 2018, 25, 353–368. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Liao, C.P.; Wang, Y.; Ramani, V.; Vandergriff, T.; Lee, M.; Patel, A.; Ariizumi, K.; Le, L.Q. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat. Commun. 2018, 9, 5014. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beert, E.; Brems, H.; Daniels, B.; De Wever, I.; Van Calenbergh, F.; Schoenaers, J.; Debiec-Rychter, M.; Gevaert, O.; De Raedt, T.; Van Den Bruel, A.; et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011, 50, 1021–1032. [Google Scholar] [CrossRef]
- Carrio, M.; Gel, B.; Terribas, E.; Zucchiatti, A.C.; Moline, T.; Rosas, I.; Teule, A.; Ramon, Y.C.S.; Lopez-Gutierrez, J.C.; Blanco, I.; et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum. Mutat. 2018, 39, 1112–1125. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Kolberg, M.; Holand, M.; Agesen, T.H.; Brekke, H.R.; Liestol, K.; Hall, K.S.; Mertens, F.; Picci, P.; Smeland, S.; Lothe, R.A. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro. Oncol. 2013, 15, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum. Genom. 2011, 5, 623–690. [Google Scholar] [CrossRef] [Green Version]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.O.; Dombi, E.; et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Invest. 2013, 123, 340–347. [Google Scholar] [CrossRef]
- Watson, A.L.; Anderson, L.K.; Greeley, A.D.; Keng, V.W.; Rahrmann, E.P.; Halfond, A.L.; Powell, N.M.; Collins, M.H.; Rizvi, T.; Moertel, C.L.; et al. Co-targeting the MAPK and PI3K/AKT/mTOR pathways in two genetically engineered mouse models of schwann cell tumors reduces tumor grade and multiplicity. Oncotarget 2014, 5, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Fischer-Huchzermeyer, S.; Chikobava, L.; Stahn, V.; Zangarini, M.; Berry, P.; Veal, G.J.; Senner, V.; Mautner, V.F.; Harder, A. Testing ATRA and MEK inhibitor PD0325901 effectiveness in a nude mouse model for human MPNST xenografts. BMC Res. Notes. 2018, 11, 520. [Google Scholar] [CrossRef]
- Peacock, J.D.; Cherba, D.; Kampfschulte, K.; Smith, M.K.; Monks, N.R.; Webb, C.P.; Steensma, M. Molecular-guided therapy predictions reveal drug resistance phenotypes and treatment alternatives in malignant peripheral nerve sheath tumors. J. Transl. Med. 2013, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Peacock, J.D.; Pridgeon, M.G.; Tovar, E.A.; Essenburg, C.J.; Bowman, M.; Madaj, Z.; Koeman, J.; Boguslawski, E.A.; Grit, J.; Dodd, R.D.; et al. Genomic Status of MET Potentiates Sensitivity to MET and MEK Inhibition in NF1-Related Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2018, 78, 3672–3687. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, S.; Ge, Y.; Tainsky, M.A. Combinatorial therapeutic targeting of BMP2 and MEK-ERK pathways in NF1-associated malignant peripheral nerve sheath tumors. Oncotarget. 2016, 7, 57171–57185. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, R.M.; den Bakker, M.A.; Dubbink, H.J.; Hop, W.C.; Dinjens, W.N.; Kros, J.M. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J. Neuropathol. Exp. Neurol. 2010, 69, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. J. Natl. Compr. Cancer Netw. 2018, 16, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, L.E.; Trievel, R.C. Structure, mechanism, and regulation of polycomb repressive complex 2. J. Biol. Chem. 2018, 293, 13805–13814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleven, A.H.; Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- He, S.; Mansour, M.R.; Zimmerman, M.W.; Ki, D.H.; Layden, H.M.; Akahane, K.; Gjini, E.; de Groh, E.D.; Perez-Atayde, A.R.; Zhu, S.; et al. Synergy between loss of NF1 and overexpression of MYCN in neuroblastoma is mediated by the GAP-related domain. Elife 2016, 5, e14713. [Google Scholar] [CrossRef]
- Ki, D.H.; He, S.; Rodig, S.; Look, A.T. Overexpression of PDGFRA cooperates with loss of NF1 and p53 to accelerate the molecular pathogenesis of malignant peripheral nerve sheath tumors. Oncogene 2017, 36, 1058–1068. [Google Scholar] [CrossRef]
- Ki, D.H.; Oppel, F.; Durbin, A.D.; Look, A.T. Mechanisms underlying synergy between DNA topoisomerase I-targeted drugs and mTOR kinase inhibitors in NF1-associated malignant peripheral nerve sheath tumors. Oncogene 2019, 38, 6585–6598. [Google Scholar] [CrossRef]
- Oppel, F.; Tao, T.; Shi, H.; Ross, K.N.; Zimmerman, M.W.; He, S.; Tong, G.; Aster, J.C.; Look, A.T. Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 2019, 15, e1008039. [Google Scholar] [CrossRef] [Green Version]
- Durbin, A.D.; Ki, D.H.; He, S.; Look, A.T. Malignant Peripheral Nerve Sheath Tumors. Adv. Exp. Med. Biol. 2016, 916, 495–530. [Google Scholar]
- Gregorian, C.; Nakashima, J.; Dry, S.M.; Nghiemphu, P.L.; Smith, K.B.; Ao, Y.; Dang, J.; Lawson, G.; Mellinghoff, I.K.; Mischel, P.S.; et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 19479–19484. [Google Scholar] [CrossRef] [Green Version]
- Keng, V.W.; Watson, A.L.; Rahrmann, E.P.; Li, H.; Tschida, B.R.; Moriarity, B.S.; Choi, K.; Rizvi, T.A.; Collins, M.H.; Wallace, M.R.; et al. Conditional Inactivation of Pten with EGFR Overexpression in Schwann Cells Models Sporadic MPNST. Sarcoma 2012, 2012, 620834. [Google Scholar] [CrossRef] [Green Version]
- Huijbregts, R.P.; Roth, K.A.; Schmidt, R.E.; Carroll, S.L. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. J. Neurosci. 2003, 23, 7269–7280. [Google Scholar] [CrossRef] [Green Version]
- Kazmi, S.J.; Byer, S.J.; Eckert, J.M.; Turk, A.N.; Huijbregts, R.P.; Brossier, N.M.; Grizzle, W.E.; Mikhail, F.M.; Roth, K.A.; Carroll, S.L. Transgenic mice overexpressing neuregulin-1 model neurofibroma-malignant peripheral nerve sheath tumor progression and implicate specific chromosomal copy number variations in tumorigenesis. Am. J. Pathol. 2013, 182, 646–667. [Google Scholar] [CrossRef] [Green Version]
- Brosius, S.N.; Turk, A.N.; Byer, S.J.; Longo, J.F.; Kappes, J.C.; Roth, K.A.; Carroll, S.L. Combinatorial therapy with tamoxifen and trifluoperazine effectively inhibits malignant peripheral nerve sheath tumor growth by targeting complementary signaling cascades. J. Neuropathol. Exp. Neurol. 2014, 73, 1078–1090. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.C.; Wu, J.; Miller, S.J.; Monk, K.R.; Shamekh, R.; Rizvi, T.A.; Decourten-Myers, G.; Vogel, K.S.; DeClue, J.E.; Ratner, N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005, 7, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Rahrmann, E.P.; Moriarity, B.S.; Otto, G.M.; Watson, A.L.; Choi, K.; Collins, M.H.; Wallace, M.; Webber, B.R.; Forster, C.L.; Rizzardi, A.E.; et al. Trp53 haploinsufficiency modifies EGFR-driven peripheral nerve sheath tumorigenesis. Am. J. Pathol. 2014, 184, 2082–2098. [Google Scholar] [CrossRef] [Green Version]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 Inactivation in Schwann Cells Produces a Severe Phenotype in the Peripheral Nervous System That Promotes the Development and Malignant Progression of Peripheral Nerve Sheath Tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Mo, W.; Chen, J.; Patel, A.; Zhang, L.; Chau, V.; Li, Y.; Cho, W.; Lim, K.; Xu, J.; Lazar, A.J.; et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell 2013, 152, 1077–1090. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, C.M.; Johnson, B.W.; Williams, S.M.; Chan, A.W.; Reczek, E.E.; Lynch, R.C.; Rioth, M.J.; McClatchey, A.; Ryeom, S.; Cichowski, K. TORC1 is essential for NF1-associated malignancies. Curr. Biol. 2008, 18, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Gonzalez-Gualda, E.; Doherty, G.J.; Munoz-Espin, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef]
- Rahrmann, E.P.; Watson, A.L.; Keng, V.W.; Choi, K.; Moriarity, B.S.; Beckmann, D.A.; Wolf, N.K.; Sarver, A.; Collins, M.H.; Moertel, C.L.; et al. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nat. Genet. 2013, 45, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Keng, V.W.; Patmore, D.M.; Kendall, J.J.; Patel, A.V.; Jousma, E.; Jessen, W.J.; Choi, K.; Tschida, B.R.; Silverstein, K.A.; et al. Insertional Mutagenesis Identifies a STAT3/Arid1b/beta-catenin Pathway Driving Neurofibroma Initiation. Cell Rep. 2016, 14, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Streubel, G.; Watson, A.; Jammula, S.G.; Scelfo, A.; Fitzpatrick, D.J.; Oliviero, G.; McCole, R.; Conway, E.; Glancy, E.; Negri, G.L.; et al. The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol. Cell 2018, 70, 371–379 e375. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse tumor model for neurofibromatosis type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Reilly, K.M.; Tuskan, R.G.; Christy, E.; Loisel, D.A.; Ledger, J.; Bronson, R.T.; Smith, C.D.; Tsang, S.; Munroe, D.J.; Jacks, T. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc. Natl. Acad. Sci. USA 2004, 101, 13008–13013. [Google Scholar] [CrossRef] [Green Version]
- Malone, C.F.; Fromm, J.A.; Maertens, O.; DeRaedt, T.; Ingraham, R.; Cichowski, K. Defining key signaling nodes and therapeutic biomarkers in NF1-mutant cancers. Cancer Discov. 2014, 4, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; McCurrach, M.E.; Braun, B.S.; De Raedt, T.; Epstein, I.; Huang, T.Q.; Lauchle, J.O.; Lee, H.; Wu, J.; Cripe, T.P.; et al. A Collaborative Model for Accelerating the Discovery and Translation of Cancer Therapies. Cancer Res. 2017, 77, 5706–5711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Ingraham, R.; Maertens, O.; Miller, A.L.; Weledji, N.; Legius, E.; Konicek, B.M.; Yan, S.C.; Graff, J.R.; Cichowski, K. Cotargeting MNK and MEK kinases induces the regression of NF1-mutant cancers. J. Clin. Invest. 2016, 126, 2181–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, R.D.; Mito, J.K.; Eward, W.C.; Chitalia, R.; Sachdeva, M.; Ma, Y.; Barretina, J.; Dodd, L.; Kirsch, D.G. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol. Cancer 2013, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Dodd, R.D.; Lee, C.L.; Overton, T.; Huang, W.; Eward, W.C.; Luo, L.; Ma, Y.; Ingram, D.R.; Torres, K.E.; Cardona, D.M.; et al. NF1(+/−) Hematopoietic Cells Accelerate Malignant Peripheral Nerve Sheath Tumor Development without Altering Chemotherapy Response. Cancer Res. 2017, 77, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, M.; Whitley, M.J.; Kuo, H.C.; Xu, E.S.; Walens, A.; Mowery, Y.M.; Van Mater, D.; Eward, W.C.; Cardona, D.M.; et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat. Commun. 2017, 8, 15999. [Google Scholar] [CrossRef]
- Fang, Y.; Elahi, A.; Denley, R.C.; Rao, P.H.; Brennan, M.F.; Jhanwar, S.C. Molecular characterization of permanent cell lines from primary, metastatic and recurrent malignant peripheral nerve sheath tumors (MPNST) with underlying neurofibromatosis-1. Anticancer Res. 2009, 29, 1255–1262. [Google Scholar]
- Sun, D.; Tainsky, M.A.; Haddad, R. Oncogene Mutation Survey in MPNST Cell Lines Enhances the Dominant Role of Hyperactive Ras in NF1 Associated Pro-Survival and Malignancy. Transl Oncogenomics 2012, 5, 1–7. [Google Scholar]
- Castellsague, J.; Gel, B.; Fernandez-Rodriguez, J.; Llatjos, R.; Blanco, I.; Benavente, Y.; Perez-Sidelnikova, D.; Garcia-Del Muro, J.; Vinals, J.M.; Vidal, A.; et al. Comprehensive establishment and characterization of orthoxenograft mouse models of malignant peripheral nerve sheath tumors for personalized medicine. Embo Mol. Med. 2015, 7, 608–627. [Google Scholar] [CrossRef]
- Pollard, K.; Banerjee, J.; Doan, X.; Wang, J.; Guo, X.; Allaway, R.; Langmead, S.; Slobogean, B.; Meyer, C.F.; Loeb, D.M. A clinically and genomically annotated nerve sheath tumor biospecimen repository. biorxiv 2020. [CrossRef]
- Rutkowski, J.L.; Kirk, C.J.; Lerner, M.A.; Tennekoon, G.I. Purification and expansion of human Schwann cells in vitro. Nat. Med. 1995, 1, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chang, L.J.; Neubauer, D.R.; Muir, D.F.; Wallace, M.R. Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab. Invest. 2016, 96, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrio, M.; Mazuelas, H.; Richaud-Patin, Y.; Gel, B.; Terribas, E.; Rosas, I.; Jimenez-Delgado, S.; Biayna, J.; Vendredy, L.; Blanco, I.; et al. Reprogramming Captures the Genetic and Tumorigenic Properties of Neurofibromatosis Type 1 Plexiform Neurofibromas. Stem. Cell Rep. 2019, 12, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobzhansky, T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 1946, 31, 269–290. [Google Scholar] [PubMed]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2020, 9, 23–38. [Google Scholar] [CrossRef]
- Semenova, G.; Stepanova, D.S.; Deyev, S.M.; Chernoff, J. Medium throughput biochemical compound screening identifies novel agents for pharmacotherapy of neurofibromatosis type 1. Biochimie 2017, 135, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, M.; Gosline, S.J.C.; Stathis, M.; Zhang, X.; Guo, X.; Guha, R.; Ryman, D.A.; Wallace, M.R.; Kasch-Semenza, L.; Hao, H.; et al. Pharmacological and genomic profiling of neurofibromatosis type 1 plexiform neurofibroma-derived schwann cells. Sci. Data 2018, 5, 180106. [Google Scholar] [CrossRef] [PubMed]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef] [PubMed]
- McCowage, G.B.; Pratilas, C.A.; Hargrave, D.R.; Moertel, C.L.; Whitlock, J.; Fox, E.; Hingorani, P.; Russo, M.W.; Dasgupta, K.; Tseng, L.; et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)–associated plexiform neurofibroma: A phase I/IIa study. J. Clin. Oncol. 2018, 36, 10504. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]
- Daud, A.; Tsai, K. Management of Treatment-Related Adverse Events with Agents Targeting the MAPK Pathway in Patients with Metastatic Melanoma. Oncologist 2017, 22, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Stalnecker, C.A.; Der, C.J. RAS, wanted dead or alive: Advances in targeting RAS mutant cancers. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.A.; Ling, B.; Ratner, N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: Reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol. Cell Biol. 1997, 17, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Dilworth, J.T.; Wojtkowiak, J.W.; Mathieu, P.; Tainsky, M.A.; Reiners, J.J., Jr.; Mattingly, R.R.; Hancock, C.N. Suppression of proliferation of two independent NF1 malignant peripheral nerve sheath tumor cell lines by the pan-ErbB inhibitor CI-1033. Cancer Biol. 2008, 7, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Wojtkowiak, J.W.; Fouad, F.; LaLonde, D.T.; Kleinman, M.D.; Gibbs, R.A.; Reiners, J.J., Jr.; Borch, R.F.; Mattingly, R.R. Induction of apoptosis in neurofibromatosis type 1 malignant peripheral nerve sheath tumor cell lines by a combination of novel farnesyl transferase inhibitors and lovastatin. J. Pharm. Exp. 2008, 326, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Widemann, B.C.; Salzer, W.L.; Arceci, R.J.; Blaney, S.M.; Fox, E.; End, D.; Gillespie, A.; Whitcomb, P.; Palumbo, J.S.; Pitney, A.; et al. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J. Clin. Oncol. 2006, 24, 507–516. [Google Scholar] [CrossRef]
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro. Oncol. 2014, 16, 707–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: A multicentre, phase 2 trial. Lancet. Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A neurofibromatosis Clinical Trials Consortium phase II study. Neuro. Oncol. 2015, 17, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Varin, J.; Poulain, L.; Hivelin, M.; Nusbaum, P.; Hubas, A.; Laurendeau, I.; Lantieri, L.; Wolkenstein, P.; Vidaud, M.; Pasmant, E.; et al. Dual mTORC1/2 inhibition induces anti-proliferative effect in NF1-associated plexiform neurofibroma and malignant peripheral nerve sheath tumor cells. Oncotarget 2016, 7, 35753–35767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.; Lu, Y.; Okuno, S.H.; Reinke, D.; Maertens, O.; Perentesis, J.; Basu, M.; Wolters, P.L.; De Raedt, T.; Chawla, S.; et al. Targeting Refractory Sarcomas and Malignant Peripheral Nerve Sheath Tumors in a Phase I/II Study of Sirolimus in Combination with Ganetespib (SARC023). Sarcoma 2020, 2020, 5784876. [Google Scholar] [CrossRef]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747. [Google Scholar] [CrossRef] [Green Version]
- Patwardhan, P.P.; Surriga, O.; Beckman, M.J.; de Stanchina, E.; Dematteo, R.P.; Tap, W.D.; Schwartz, G.K. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin. Cancer Res. 2014, 20, 3146–3158. [Google Scholar] [CrossRef] [Green Version]
- Kolberg, M.; Bruun, J.; Murumagi, A.; Mpindi, J.P.; Bergsland, C.H.; Holand, M.; Eilertsen, I.A.; Danielsen, S.A.; Kallioniemi, O.; Lothe, R.A. Drug sensitivity and resistance testing identifies PLK1 inhibitors and gemcitabine as potent drugs for malignant peripheral nerve sheath tumors. Mol. Oncol. 2017, 11, 1156–1171. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Moreno Samayoa, A.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A is an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Clin. Cancer Res. 2020. [CrossRef] [Green Version]
- Wojcik, J.B.; Marchione, D.M.; Sidoli, S.; Djedid, A.; Lisby, A.; Majewski, J.; Garcia, B.A. Epigenomic reordering induced by Polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankowski, K.J.; Wang, C.; Patnaik, S.; Schoenen, F.J.; Southall, N.; Li, D.; Teper, Y.; Sun, W.; Kandela, I.; Hu, D.; et al. Metarrestin, a perinucleolar compartment inhibitor, effectively suppresses metastasis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, J.S.; Musi, E.; Schwartz, G.K. Selinexor (KPT-330) Induces Tumor Suppression through Nuclear Sequestration of IkappaB and Downregulation of Survivin. Clin. Cancer Res. 2017, 23, 4301–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahller, Y.Y.; Rangwala, F.; Ratner, N.; Cripe, T.P. Malignant peripheral nerve sheath tumors with high and low Ras-GTP are permissive for oncolytic herpes simplex virus mutants. Pediatr Blood Cancer. 2006, 46, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Farassati, F.; Pan, W.; Yamoutpour, F.; Henke, S.; Piedra, M.; Frahm, S.; Al-Tawil, S.; Mangrum, W.I.; Parada, L.F.; Rabkin, S.D.; et al. Ras signaling influences permissiveness of malignant peripheral nerve sheath tumor cells to oncolytic herpes. Am. J. Pathol. 2008, 173, 1861–1872. [Google Scholar] [CrossRef] [Green Version]
- Antoszczyk, S.; Spyra, M.; Mautner, V.F.; Kurtz, A.; Stemmer-Rachamimov, A.O.; Martuza, R.L.; Rabkin, S.D. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014, 16, 1057–1066. [Google Scholar] [CrossRef] [Green Version]
- Deyle, D.R.; Escobar, D.Z.; Peng, K.W.; Babovic-Vuksanovic, D. Oncolytic measles virus as a novel therapy for malignant peripheral nerve sheath tumors. Genes 2015, 565, 140–145. [Google Scholar] [CrossRef]
- Jackson, J.D.; Markert, J.M.; Li, L.; Carroll, S.L.; Cassady, K.A. STAT1 and NF-kappaB Inhibitors Diminish Basal Interferon-Stimulated Gene Expression and Improve the Productive Infection of Oncolytic HSV in MPNST Cells. Mol. Cancer Res. 2016, 14, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Currier, M.A.; Sprague, L.; Rizvi, T.A.; Nartker, B.; Chen, C.Y.; Wang, P.Y.; Hutzen, B.J.; Franczek, M.R.; Patel, A.V.; Chaney, K.E.; et al. Aurora A kinase inhibition enhances oncolytic herpes virotherapy through cytotoxic synergy and innate cellular immune modulation. Oncotarget 2017, 8, 17412–17427. [Google Scholar] [CrossRef] [Green Version]
- Ghonime, M.G.; Cassady, K.A. Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol. Res. 2018, 6, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.A.; Nalepa, G.; Yang, F.C.; Bowers, D.C.; Ho, C.Y.; Hutchins, G.D.; Croop, J.M.; Vik, T.A.; Denne, S.C.; Parada, L.F.; et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: A phase 2 trial. Lancet. Oncol. 2012, 13, 1218–1224. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Grovola, M.R.; Xie, H.; Coggins, G.E.; Duggan, P.; Hasan, R.; Huang, J.; Lin, D.W.; Song, C.; Witek, G.M.; et al. Comprehensive pharmacological profiling of neurofibromatosis cell lines. Am. J. Cancer Res. 2017, 7, 923–934. [Google Scholar] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, K.B.; Largaespada, D.A. New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes 2020, 11, 477. https://doi.org/10.3390/genes11050477
Williams KB, Largaespada DA. New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes. 2020; 11(5):477. https://doi.org/10.3390/genes11050477
Chicago/Turabian StyleWilliams, Kyle B., and David A. Largaespada. 2020. "New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors" Genes 11, no. 5: 477. https://doi.org/10.3390/genes11050477
APA StyleWilliams, K. B., & Largaespada, D. A. (2020). New Model Systems and the Development of Targeted Therapies for the Treatment of Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes, 11(5), 477. https://doi.org/10.3390/genes11050477