Identification of Molecular Mechanisms Related to Pig Fatness at the Transcriptome and miRNAome Levels

 , ,
, ,  , , , and
, , , and
Abstract
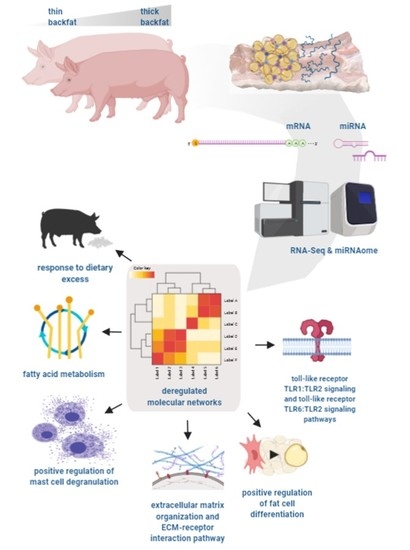
:
1. Introduction
2. Materials and Methods
2.1. Animals, Phenotype Data Collection, and Tissue Sampling
2.2. Whole Transcriptome Sequencing (RNA-seq)
2.3. MicroRNA Sequencing
2.4. Validation of NGS Results
3. Results
3.1. RNA Sequencing Results—Differentially-Expressed Genes
3.2. MiRNA Sequencing Results—Differentially-Expressed miRNAs
3.3. Analysis of Pathways Common for DEGs and DE miRNAs
3.4. Validation of Obtained Data
4. Discussion
Enriched Metabolic Process and Pathways Associated with Fat Deposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Hussain, Z.; Khan, J.A. Food intake regulation by leptin: Mechanisms mediating gluconeogenesis and energy expenditure. Asian Pac. J. Trop. Med. 2017, 10, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Elmquist, J.K.; Elias, C.F.; Saper, C.B. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron 1999, 22, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.; Oliveira, T.; Fernandes, R. Biochemistry of adipose tissue: An endocrine organ. Arch. Med. Sci. 2013, 9, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Switonski, M.; Stachowiak, M.; Cieslak, J.; Bartz, M.; Grzes, M. Genetics of fat tissue accumulation in pigs: A comparative approach. J. Appl. Genet. 2010, 51, 153–168. [Google Scholar] [CrossRef]
- Allison, D.B.; Neale, M.C.; Kezis, M.I.; Alfonso, V.C.; Heshka, S.; Heymsfield, S.B. Assortative mating for relative weight: Genetic implications. Behav. Genet. 1996, 26, 103–111. [Google Scholar] [CrossRef]
- Ashrafi, K. Obesity and the Regulation of Fat Metabolism. WormBook. 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK19757/ (accessed on 28 May 2020).
- Ponsuksili, S.; Murani, E.; Brand, B.; Schwerin, M.; Wimmers, K. Integrating expression profiling and whole-genome association for dissection of fat traits in a porcine model. J. Lipid Res. 2011, 52, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Qiao, R.; Gao, J.; Zhang, Z.; Li, L.; Xie, X.; Fan, Y.; Cui, L.; Ma, J.; Ai, H.; Ren, J.; et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet. Sel. Evol. 2015, 47, 17. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel. Evol. 2017, 49, 21. [Google Scholar] [CrossRef] [Green Version]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. DMM Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausman, D.B.; Hausman, G.J.; Martin, R.J. Influence of the pituitary on lipolysis and lipogenesis in fetal pig adipose tissue. Horm. Metab. Res. 1993, 25, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Montarelo, D.; Madsen, O.; Alves, E.; Rodríguez, M.C.; Folch, J.M.; Noguera, J.L.; Groenen, M.A.M.; Fernández, A.I. Identification of genes regulating growth and fatness traits in pig through hypothalamic transcriptome analysis. Physiol. Genom. 2014, 46, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Wu, Q.; Li, Y.; Shang, H.; Guo, K.; Wu, J.; Wei, H.; Zhao, J.; Yu, J.; Li, M.H. Transcriptome profiling identifies differentially expressed genes in postnatal developing pituitary gland of miniature pig. DNA Res. 2014, 21, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Piórkowska, K.; Żukowski, K.; Tyra, M.; Szyndler-Nędza, M.; Szulc, K.; Skrzypczak, E.; Ropka-Molik, K. The pituitary transcriptional response related to feed conversion in pigs. Genes 2019, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.A.; Clarke, I.J. Adipose Tissue Hormones and the Regulation of Food Intake. J. Neuroendocrinol. 2008, 20, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Havel, P.J. Role of adipose tissue in body-weight regulation: Mechanisms regulating leptin production and energy balance. Proc. Nutr. Soc. 2000, 59, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Qi, X.; Hu, M.; Lin, R.; Hou, Y.; Wang, Z.; Zhou, H.; Zhao, Y.; Luan, Y.; Zhao, S.; et al. Transcriptome analysis of adipose tissue indicates that the cAMP signaling pathway affects the feed efficiency of pigs. Genes 2018, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Benítez, R.; Trakooljul, N.; Núñez, Y.; Isabel, B.; Murani, E.; De Mercado, E.; Gómez-Izquierdo, E.; García-Casco, J.; López-Bote, C.; Wimmers, K.; et al. Breed, diet, and interaction effects on adipose tissue transcriptome in iberian and duroc pigs fed different energy sources. Genes 2019, 10, 589. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, A.; Óvilo, C.; Núñez, Y.; Benítez, R.; López-Garcia, A.; García, F.; Félix, M.D.R.; Laranjo, M.; Charneca, R.; Martins, J.M. Comparative transcriptomic analysis of subcutaneous adipose tissue from local pig breeds. Genes 2020, 11, 422. [Google Scholar] [CrossRef] [Green Version]
- Xing, K.; Zhu, F.; Zhai, L.; Chen, S.; Tan, Z.; Sun, Y.; Hou, Z.; Wang, C. Identification of genes for controlling swine adipose deposition by integrating transcriptome, whole-genome resequencing, and quantitative trait loci data. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ropka-Molik, K.; Dusik, A.; Piórkowska, K.; Tyra, M.; Oczkowicz, M.; Szmatoła, T. Polymorphisms of the membrane-associated ring finger 4, ubiquitin protein ligase gene (MARCH4) and its relationship with porcine production traits. Livest. Sci. 2015, 178, 18–26. [Google Scholar] [CrossRef]
- Tyra, M.; Żak, G. Analysis of the possibility of improving the indicators of pork quality through selection with particular consideration of intramuscular fat (imf) conntenttent*. Ann. Anim. Sci. 2013, 13, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 May 2020).
- Stocks, M.B.; Moxon, S.; Mapleson, D.; Woolfenden, H.C.; Mohorianu, I.; Folkes, L.; Schwach, F.; Dalmay, T.; Moulton, V. The UEA sRNA workbench: A suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 2012, 28, 2059–2061. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.; van Dongen, S.; Enright, A. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Urgese, G.; Paciello, G.; Acquaviva, A.; Ficarra, E. IsomiR-SEA: An RNA-Seq analysis tool for miRNAs/isomiRs expression level profiling and miRNA-mRNA interaction sites evaluation. BMC Bioinformatics 2016, 17, 148. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.-L.; Maniou, S.; Karathanou, K.; Kalfakakou, D. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Piórkowska, K.; Oczkowicz, M.; Rózycki, M.; Ropka-Molik, K.; Kajtoch, A.P.K. Novel porcine housekeeping genes for real-time RT-PCR experiments normalization in adipose tissue: Assessment of leptin mRNA quantity in different pig breeds. Meat Sci. 2011, 87, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Piórkowska, K.; Tyra, M.; Ropka-Molik, K.; Podbielska, A. Evolution of peroxisomal trans-2-enoyl-CoA reductase (PECR) as candidate gene for meat quality. Livest. Sci. 2017, 201, 85–91. [Google Scholar] [CrossRef]
- Jacyno, E.; Pietruszka, A.; Kawecka, M.; Biel, W.; Kołodziej-Skalska, A. Phenotypic correlations of backfat thickness with meatiness traits, intramuscular fat, Longissimus muscle cholesterol and fatty acid composition in pigs. South Afr. J. Anim. Sci. 2015, 45, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E28. [Google Scholar] [CrossRef] [Green Version]
- Hulver, M.W.; Berggren, J.R.; Carper, M.J.; Miyazaki, M.; Ntambi, J.M.; Hoffman, E.P.; Thyfault, J.P.; Stevens, R.; Dohm, G.L.; Houmard, J.A.; et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005, 2, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Flowers, M.T.; Ntambi, J.M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 2008, 19, 248–256. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Miyazaki, M.; Socci, N.D.; Hagge-Greenberg, A.; Liedtke, W.; Soukas, A.A.; Sharma, R.; Hudgins, L.C.; Ntambi, J.M.; Friedman, J.M. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 2002, 297, 240–243. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, W.; Zhang, Z.; Fan, Y.; Xie, X.; Ai, H.; Ma, J.; Xiao, S.; Huang, L.; Ren, J. Genome-Wide Association Analyses for Fatty Acid Composition in Porcine Muscle and Abdominal Fat Tissues. PLoS ONE 2013, 8, e65554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig-Oliveras, A.; Revilla, M.; Castelló, A.; Fernández, A.I.; Folch, J.M.; Ballester, M. Expression-based GWAS identifies variants, gene interactions and key regulators affecting intramuscular fatty acid content and composition in porcine meat. Sci. Rep. 2016, 6, 31803. [Google Scholar] [CrossRef] [PubMed]
- Matoušková, P.; Hanousková, B.; Skálová, L. Micrornas as potential regulators of glutathione peroxidases expression and their role in obesity and related pathologies. Int. J. Mol. Sci. 2018, 19, 1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Wu, Y.; Wang, J.; Chen, J.; Huang, Y.; Rao, J.; Feng, C. MicroRNA-24 promotes 3T3-L1 adipocyte differentiation by directly targeting the MAPK7 signaling. Biochem. Biophys. Res. Commun. 2016, 474, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Piórkowska, K.; Małopolska, M.; Ropka-Molik, K.; Nędza, M.S.; Wiechniak, A.; Żukowski, K.; Lambert, B.; Tyra, M. Evaluation of scd, acaca and fasn mutations: Effects on pork quality and other production traits in pigs selected based on RNA-seq results. Animals 2020, 10, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, R.N.; Noguera, J.L.; García-Santana, M.J.; González, E.; Tejeda, J.F.; Ros-Freixedes, R.; Ibáñez-Escriche, N. Five genomic regions have a major impact on fat composition in Iberian pigs. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Alves, E.; Corominas, J.; Folch, J.M.; Casellas, J.; Noguera, J.L.; Silió, L.; Fernández, A.I. Survey of SSC12 regions affecting fatty acid composition of intramuscular fat using high-density SNP data. Front. Genet. 2012, 2, 101. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-W.; Choi, Y.-I.; Choi, J.-S.; Kim, J.-J.; Choi, B.-H.; Kim, T.-H.; Kim, K.-S. Porcine Fatty Acid Synthase Gene Polymorphisms Are Associated with Meat Quality and Fatty Acid Composition. Korean J. Food Sci. Anim. Resour. 2011, 31, 356–365. [Google Scholar] [CrossRef]
- Grzes, M.; Sadkowski, S.; Rzewuska, K.; Szydlowski, M.; Switonski, M. Pig fatness in relation to FASN and INSIG2 genes polymorphism and their transcript level. Mol. Biol. Rep. 2016, 43, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Brennan, A.M.; Mantzoros, C.S. Drug Insight: The role of leptin in human physiology and pathophysiology—Emerging clinical applications. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 318–327. [Google Scholar] [CrossRef]
- Lönnqvist, F.; Arner, P.; Nordfors, L.; Schalling, M. Overexpression of the obese (ob) gene in adipose tissue of human obese subjects. Nat. Med. 1995, 1, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M. Leptin: Fights against obesity! Pak. J. Physiol. 2006, 2, 54–60. [Google Scholar]
- Hill, J.O.; Wyatt, H.R.; Peters, J.C. Energy balance and obesity. Circulation 2012, 126, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ridderstråle, M.; Johansson, L.E.; Rastam, L.; Lindblad, U. Increased risk of obesity associated with the variant allele of the PPARGC1A Gly482Ser polymorphism in physically inactive elderly men. Diabetologia 2006, 49, 496–500. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhao, Y.; Tang, N.; Feng, R.; Yang, X.; Lu, N.; Wen, J.; Li, L. Pax6 Directly Down-Regulates Pcsk1n Expression Thereby Regulating PC1/3 Dependent Proinsulin Processing. PLoS ONE 2012, 7, e46934. [Google Scholar] [CrossRef]
- Shi, M.A.; Shi, G.P. Different roles of mast cells in obesity and diabetes: Lessons from experimental animals and humans. Front. Immunol. 2012, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Finlin, B.S.; Confides, A.L.; Zhu, B.; Boulanger, M.C.; Memetimin, H.; Taylor, K.W.; Johnson, Z.R.; Westgate, P.M.; Dupont-Versteegden, E.E.; Kern, P.A. Adipose Tissue Mast Cells Promote Human Adipose Beiging in Response to Cold. Sci. Rep. 2019, 9, 8658. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.; Chun, T.H.; Kang, L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem. Pharmacol. 2016, 119, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yan, Y.; Xv, W.; Qian, G.; Li, C.; Zou, H.; Li, Y. A New Insight into the Roles of MiRNAs in Metabolic Syndrome. BioMed Res. Int. 2018. [Google Scholar] [CrossRef] [Green Version]
- Heneghan, H.M.; Miller, N.; McAnena, O.J.; O’brien, T.; Kerin, M.J. Differential miRNA expression in omental adipose tissue and in the circulation of obese patients identifies novel metabolic biomarkers. J. Clin. Endocrinol. Metab. 2011, 96, E846–E850. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Tordjman, J.; Le Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Le Guerre-Millo, M.; Poitou, C.; Zucker, J.-D.; et al. Fibrosis in Human Adipose Tissue: Composition, Distribution, and Link With Lipid Metabolism and Fat Mass Loss. Am. Diabetes Assoc. 2010, 59, 2817–2825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrinelli, V.; Heuvingh, J.; Du Roure, O.; Rouault, C.; Devulder, A.; Klein, C.; Lacasa, M.; Clément, E.; Lacasa, D.; Clément, K. Human adipocyte function is impacted by mechanical cues. J. Pathol. 2014, 233, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Al-Mass, A.; Atizado, V.; Al-Hubail, A.; Al-Ghimlas, F.; Al-Arouj, M.; Bennakhi, A.; Dermime, S.; Behbehani, K. Elevated expression of the toll like receptors 2 and 4 in obese individuals: Its significance for obesity-induced inflammation. J. Inflamm. 2012, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef]
- Deiuliis, J. MicroRNAs as regulators of metabolic disease: Pathophysiologic significance and emerging role as biomarkers and therapeutics. Int. J. Obes. Vol. 2016, 40, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Tanaka, M.; Yamakage, H.; Metabolism, Y.S.; Satoh-Asahara, N. Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism 2015, 64, 1490–1499. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Backfat Thickness (cm) | Weight of Peritoneal Fat (kg) | |||||||
|---|---|---|---|---|---|---|---|---|
| L | H | L | H | |||||
| Pietrain | 0.71 | ±0.0.1 b | 1.29 | ±0.10 a | 0.15 | ±0.001 b | 0.31 | ±0.051 a |
| Hampshire | 0.99 | ±0.03 b | 1.54 | ±0.20 a | 0.30 | ±0.016 | 0.29 | ±0.067 |
| Large White | 1.04 | ±0.06 | 1.40 | ±0.14 | 0.23 | ±0.070 | 0.31 | ±0.122 |
| Gene Ontology/Accession Number | FDR * | N | Gene Name |
|---|---|---|---|
| Extracellular matrix organization (GO:0030198) | 9.2 × 10−3 | 6 | CYR61; ELN; GFAP; HPSE; POSTN; VTN |
| Positive regulation of mast cell degranulation (GO:0043306) | 2.4 × 10−3 | 4 | FGR; FCER1A; FCER1G; ZAP70 |
| Cell adhesion (GO:0007155) | 3.5 × 10−3 | 12 | TNFAIP6; WISP1; CTGF; CYR61; HAS1; LYVE1; NOV; POSTN; SELL; TNC; THBS2; THBS3 |
| Innate immune response (GO:0045087) | 5.9 × 10−2 | 14 | FGR; FCER1G; MX1; MX2; S100A8; B2M; BST2; FGB; IFIH1; JCHAIN; LCN2; PML; TLR2; ZAP70; |
| Fatty acid biosynthetic process (GO:0006633) | 8.4 × 10−3 | 4 | ACACA; SCD; FASN; ACLY |
| Positive regulation of apoptotic process (GO:0043065) | 5.6 × 10−3 | 10 | BMF; ALDH1A2; CLU; CYP1B1; GADD45G; IGFBP3; SFRP2; TOP2A; TGM2; ZBTB16 |
| Long-chain fatty acid biosynthetic process (GO:0042759) | 6.0 × 10−3 | 3 | PLPP1; SCD; SCD5 |
| Response to dietary excess (GO:0002021) | 8.6 × 10−3 | 3 | PPARGC1A; LEP; PCSK1N |
| Positive regulation of fat cell differentiation (GO:0045600) | 6.8 × 10−3 | 5 | CCDC3; MEDAG; SFRP2; ZBTB16; ZNF385A |
| Inflammatory response (GO:0006954) | 8.6 × 10−3 | 11 | CCL5; CCR5; CD180; FAS; CXCL10; C5AR1; PLP1; TSPAN2; TBXA2R; TLR2; ZAP70 |
| Gene | Accession Number | Pietrain | Large White | Hampshire | |||
|---|---|---|---|---|---|---|---|
| FC | FDR | FC | FDR | FC | FDR | ||
| LEP | ENSSSCG00000040464 | 1.41 | 0.03 | 2.14 | 0.001 | 1.10 | ns |
| ACACA | ENSSSCG00000017694 | 1.62 | 0.03 | 1.67 | 0.001 | 1.52 | ns |
| SCD | ENSSSCG00000010554 | 2.95 | 0.001 | 1.66 | 0.001 | 2.23 | 0.04 |
| SCD5 | ENSSSCG00000009245 | −1.32 | 0.04 | −1.09 | ns | −2.28 | 0.001 |
| FASN | ENSSSCG00000029944 | 1.58 | ns | 1.47 | 0.02 | 3.07 | 0.005 |
| ACOX3 | ENSSSCG00000008724 | 1.40 | 0.05 | 1.32 | ns | 2.06 | 0.001 |
| C2 | ENSSSCG00000001422 | −3.18 | 0.001 | −2.00 | 0.001 | 2.31 | 0.01 |
| ACLY | ENSSSCG00000017421 | 1.65 | 0.05 | 2.83 | 0.001 | 1.13 | ns |
| TNC | ENSSSCG00000005494 | −3.23 | 0.0004 | −2.55 | 0.001 | −4.17 | ns |
| PPARGC1A | ENSSSCG00000029275 | 1.16 | ns | 13.45 | 0.0001 | 3.27 | 0.05 |
| PCSK1N | ENSSSCG00000021328 | 1.07 | 0.05 | −1.52 | ns | −2.99 | 0.05 |
| TLR2 | ENSSSCG00000009002 | −2.63 | 0.001 | −1.59 | 0.05 | −1.34 | ns |
| FGR | ENSSSCG00000003578 | 1.13 | 0.05 | 1.62 | 0.001 | −1.10 | ns |
| FCER1A | ENSSSCG00000006413 | 1.40 | 0.001 | −1.19 | 0.01 | −2.69 | ns |
| FCER1G | ENSSSCG00000006357 | −2.40 | 0.001 | −1.40 | 0.05 | −1.22 | ns |
| Gene Ontology/Accession Number | FDR * | N Target Genes | N miRNAs | miRNAs |
|---|---|---|---|---|
| Extracellular matrix organization (GO:0030198) | <1.0 × 10−325 | 113 | 5 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; hsa-miR-145-5p; hsa-let-7d-5p |
| Extracellular matrix disassembly (GO:0022617) | <1.0 × 10−325 | 46 | 6 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; hsa-miR-145-5p; hsa-let-7d-5p |
| Cellular lipid metabolic process (GO:0044255) | 3.1 × 10−13 | 63 | 8 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; has-miR-143-3p; has-miR-142-5p; has-miR-145-5p; hsa-let-7d-5p |
| Cell junction organization (GO:0034330) | <1.0 × 10−325 | 66 | 6 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-143-3p; hsa-miR-145-5p; hsa-let-7d-5p |
| MyD88-independent toll-like receptor signaling pathway (GO:0002756) | <1.0 × 10−325 | 44 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; hsa-miR-145-5p; hsa-let-7d-5p; has-miR-378a-3p |
| Cellular component disassembly involved in execution phase of apoptosis (GO:0006921) | <1.0 × 10−325 | 28 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; hsa-miR-145-5p; hsa-miR-145-5p; hsa-miR-378a-3p |
| Toll-like receptor TLR1:TLR2 signaling pathway (GO:0038123)Toll-like receptor TLR6:TLR2 signaling pathway (GO:0038124) | <1.0 × 10−325 | 39 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; hsa-miR-139-5p; has-miR-378a-3p; hsa-let-7d-5p |
| Innate immune response (GO:0045087) | <1.0 × 10−325 | 213 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-24-3p; has-miR-26a-5p; hsa-miR-139-5p; has-miR-142-5p; has-miR-145-5p; hsa-let-7d-5p |
| DE miRNAs | DEGs | |||||||
|---|---|---|---|---|---|---|---|---|
| Pathways | FDR * | N | miRNAs | N | N Target Genes | FDR* | N | Genes |
| Fatty acid metabolism (hsa01212/ssc01212) | 6.6 × 10−16 | 4 | hsa-miR-100-5p; hsa-miR-10b-5p; hsa-miR-143-3p; hsa-miR-24-3p | 9 | FASN; ACACA;MCAT; ACAA2; ACAA1; CPT1A; CPT2; SCD; ELOVL5 | 5.4 × 10−3 | 5 | ACACA;ACOX3; FASN; SCD5; SCD |
| ECM−receptor interaction (hsa04512/ssc04512) | <1.0 × 10−325 | 4 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-143-3p; hsa-miR-145-5p | 35 | SPP1; CD44; LAMC1; LAMC3; ITGA3; ITGA5; FN1; TNC; COL1A2; COL4A2; COL5A1; COL5A2; COL6A1; COL27A1; ITGB1; GP5; THBS1 | 2.4 × 10−2 | 11 | CHAD; COL1A1; COL5A3; COL6A6; COL11A1; LAMC2; TNC; THBS2; THBS3; THBS4; VTN |
| Hippo-signaling pathway (hsa04390/ssc04390) | <1.0 × 10−325 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-145-5p; hsa-miR-24-3p; hsa-miR-139-5p; hsa-let-7d-5p; hsa-miR-26a-5p | 87 | PPP2CA; BMP5; BMP2; BMP7; FGF1; ACTG1; PPP1CB; CTGF; Figure S2 | 0.01 | 7 | YWHAH; CTGF; FZD2; FZD4; NKD1; TGFB3; WNT2B |
| Fatty acid biosynthesis (hsa00061/ssc00061) | <1.0 × 10−325 | 4 | hsa-miR-100-5p; hsa-miR-10b-5p; hsa-miR-143-3p; hsa-miR-24-3p | 3 | FASN; ACACA; MCAT | ns | 2 | FASN; ACACA |
| Cell cycle (hsa04110/ssc04110) | <1.0 × 10−325 | 9 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-10b-5p; hsa-miR-143-3p; hsa-miR-24-3p; hsa-miR-26a-5p; hsa-miR-142-5p; hsa-let-7d-5p | 78 | ESPL1; CDC6; GSK3B; CCNB2; RBL2; PCNA; E2F1E2F2;YWHAE; MCM6; MCM2 Figure S3 | 0.003 | 5 | E2F2; GADD45G; MCM2; TGFB3; YWHAH; |
| P53-signaling pathways (hsa04115/ ssc04151) | 2.5 × 10−9 | 7 | hsa-let-7a-5p; hsa-let-7i-5p; hsa-miR-26a-5p; hsa-miR-143-3p; hsa-miR-10b-5p; hsa-miR-142-5p; hsa-miR-378-3p | 60 | ZMAT3; CCNB2; CCNB1; CDK4; BID; THBS1; CDK2 CCND2; PERP; RRM2B; CDK1; CDKN2A; CDK6 CHEK1; TP53; APAF1; PMAIP1; CD82; CASP3 | 0.0002 | 13 | CHAD; COL1A1; COL6A6; LAMC2; PCK1; THBS2; THBS3; THBS4; TLR2; TNC; VTN; YWHAH |
| DEGs | Correlation | miRNAs | Correlation |
|---|---|---|---|
| ROCK1 | 0.56 | hsa-miR-26a-5p | 0.81 * |
| LRP12 | 0.66 * | hsa-let-7a-5p | 0.50 |
| ACACA | 0.91 ** | hsa-mir-100-5p | 0.75 * |
| HK2 | 0.87 ** | hsa-mir-378a-3p | 0.40 |
| LRP6 | 0.75 * | hsa-mir-103a-3p | 0.88 * |
| LEP | 0.94 ** | hsa-miR-125b-5p | 0.73 |
| TNC | 0.83 * | ||
| PCK1 | 0.95 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ropka-Molik, K.; Pawlina-Tyszko, K.; Żukowski, K.; Tyra, M.; Derebecka, N.; Wesoły, J.; Szmatoła, T.; Piórkowska, K. Identification of Molecular Mechanisms Related to Pig Fatness at the Transcriptome and miRNAome Levels. Genes 2020, 11, 600. https://doi.org/10.3390/genes11060600
Ropka-Molik K, Pawlina-Tyszko K, Żukowski K, Tyra M, Derebecka N, Wesoły J, Szmatoła T, Piórkowska K. Identification of Molecular Mechanisms Related to Pig Fatness at the Transcriptome and miRNAome Levels. Genes. 2020; 11(6):600. https://doi.org/10.3390/genes11060600
Chicago/Turabian StyleRopka-Molik, Katarzyna, Klaudia Pawlina-Tyszko, Kacper Żukowski, Mirosław Tyra, Natalia Derebecka, Joanna Wesoły, Tomasz Szmatoła, and Katarzyna Piórkowska. 2020. "Identification of Molecular Mechanisms Related to Pig Fatness at the Transcriptome and miRNAome Levels" Genes 11, no. 6: 600. https://doi.org/10.3390/genes11060600
APA StyleRopka-Molik, K., Pawlina-Tyszko, K., Żukowski, K., Tyra, M., Derebecka, N., Wesoły, J., Szmatoła, T., & Piórkowska, K. (2020). Identification of Molecular Mechanisms Related to Pig Fatness at the Transcriptome and miRNAome Levels. Genes, 11(6), 600. https://doi.org/10.3390/genes11060600