A Missense Variant in ALDH5A1 Associated with Canine Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD) in the Saluki Dog

 , , ,
, , ,  , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
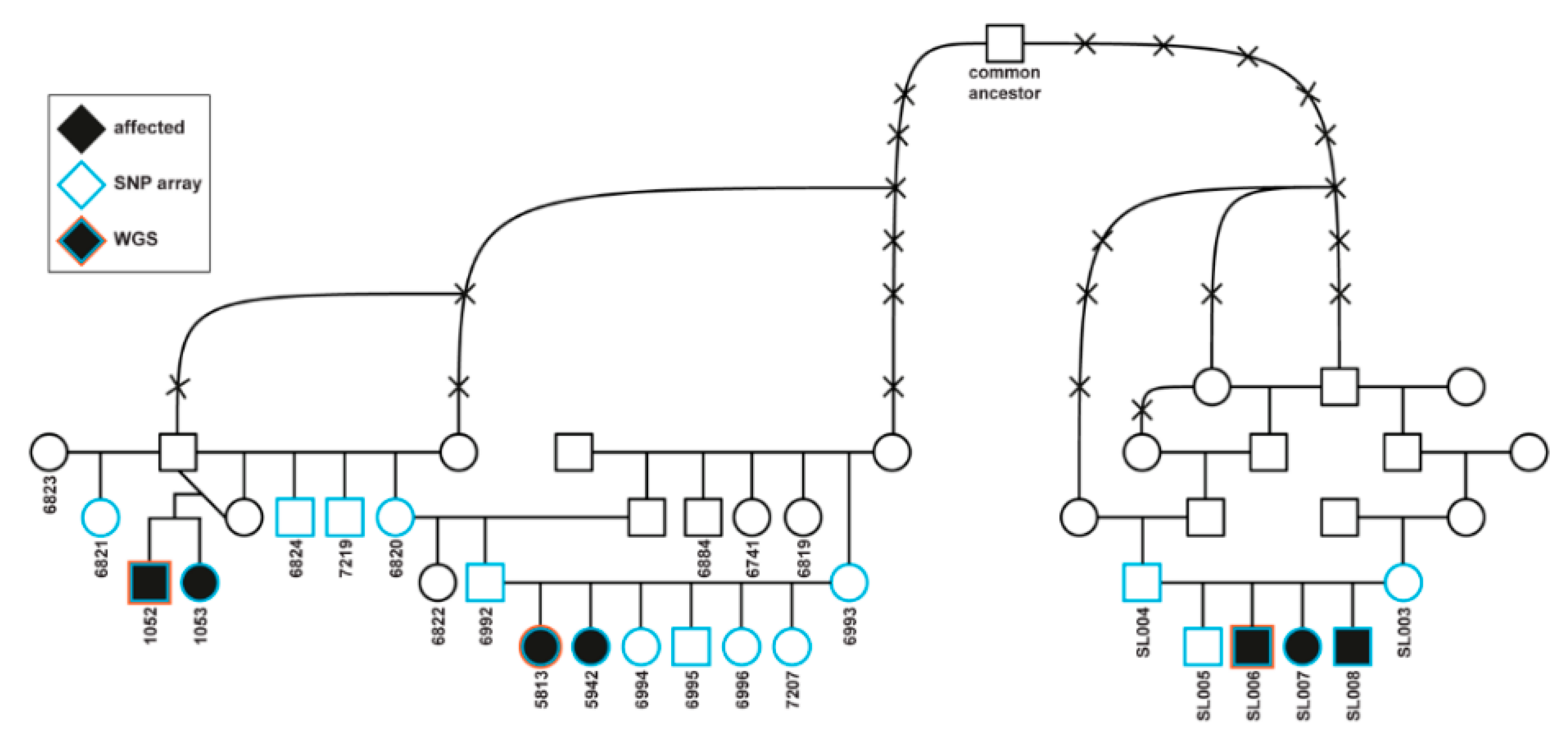
2.1. Affected Dogs
2.2. Control Dogs
2.2.1. MRI Evaluation
2.2.2. Targeted Metabolic Testing
2.3. Affected Saluki Dogs
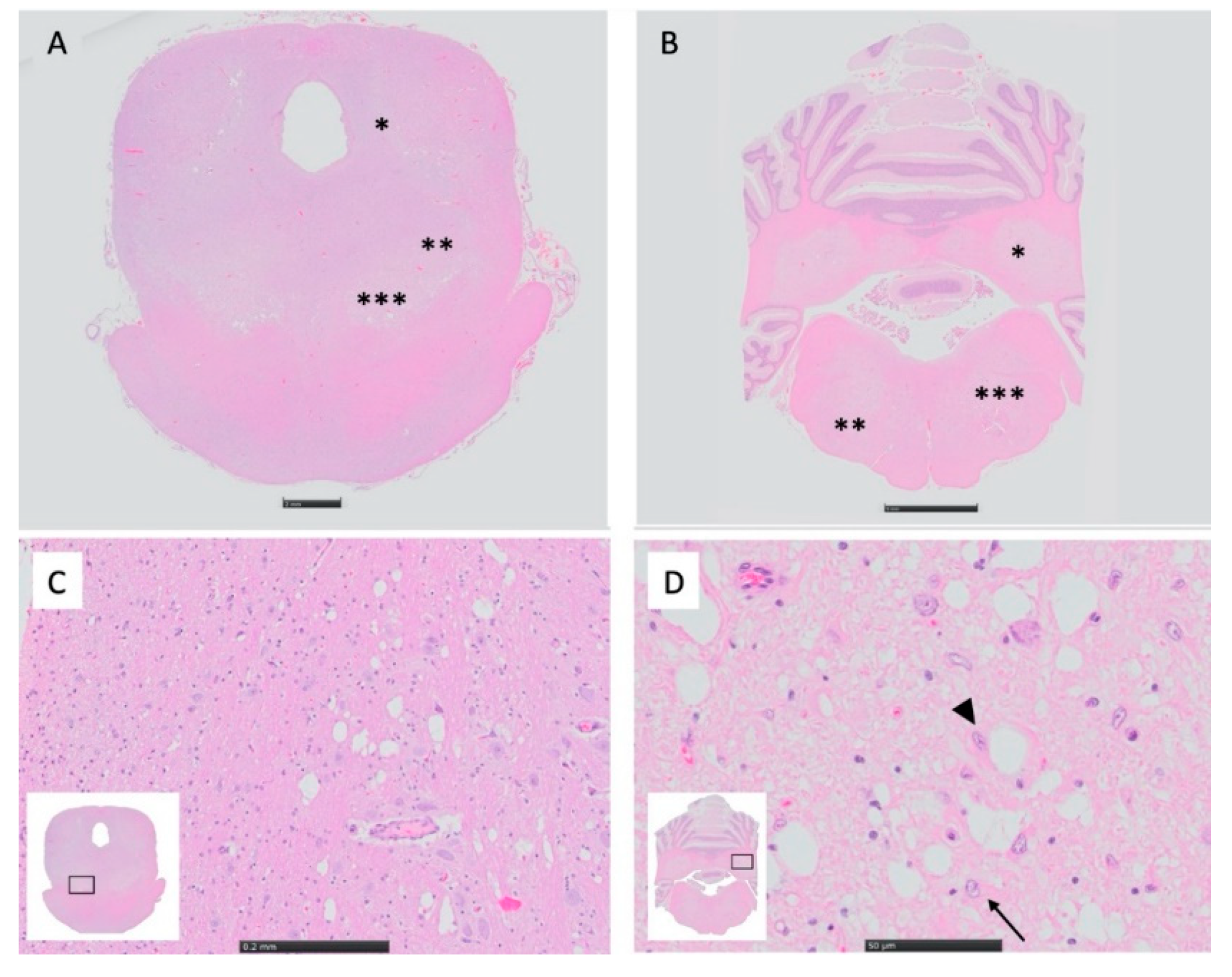
MRI and Histopathology
2.4. Sample Collection and DNA Extraction
2.5. Genome-Wide Association Scan
2.6. Whole-Genome Sequence
2.7. Genotyping
2.8. RT-PCR
2.9. Targeted Metabolic Testing
3. Results
3.1. Affected Dogs
3.2. Clinical Phenotype
3.3. MRI and Histopathology
3.4. Genetic Analysis
3.5. Targeted Metabolic Testing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- El-Hattab, A.W. Inborn errors of metabolism. Clin. Perinatol. 2015, 42, 413–439. [Google Scholar] [CrossRef]
- Seijo-Martinez, M.; Navarro, C.; Castro del Rio, M.; Vila, O.; Puig, M.; Ribes, A.; Butron, M. L-2-hydroxyglutaric aciduria: Clinical, neuroimaging, and neuropathological findings. Arch. Neurol. 2005, 62, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Jolly, R.D.; Walkley, S.U. Lysosomal storage diseases of animals: An essay in comparative pathology. Vet. Pathol. 1997, 34, 527–548. [Google Scholar] [CrossRef] [Green Version]
- Mansour, T.A.; Woolard, K.D.; Vernau, K.L.; Ancona, D.M.; Thomasy, S.M.; Sebbag, L.; Moore, B.A.; Knipe, M.F.; Seada, H.A.; Cowan, T.M.; et al. Whole genome sequencing for mutation discovery in a single case of lysosomal storage disease (MPS type 1) in the dog. Sci. Rep. 2020, 10, 6558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucot, K.L.; Dickinson, P.J.; Finno, C.J.; Mansour, T.A.; Letko, A.; Minor, K.M.; Mickelson, J.R.; Drogemuller, C.; Brown, C.T.; Bannasch, D.L. A Missense Mutation in the Vacuolar Protein Sorting 11 (VPS11) Gene Is Associated with Neuroaxonal Dystrophy in Rottweiler Dogs. G3 Genes Genomes Genet. 2018, 8, 2773–2780. [Google Scholar] [CrossRef] [Green Version]
- Haskins, M.E.; Giger, U.; Patterson, D.F. Animal models of lysosomal storage diseases: Their development and clinical relevance. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Kamboj, M. Clinical approach to the diagnoses of inborn errors of metabolism. Pediatr. Clin. N. Am. 2008, 55, 1113–1127. [Google Scholar] [CrossRef]
- Saudubray, J.M.; Garcia-Cazorla, A. An overview of inborn errors of metabolism affecting the brain: From neurodevelopment to neurodegenerative disorders. Dialogues Clin. Neurosci. 2018, 20, 301–325. [Google Scholar] [CrossRef] [Green Version]
- Sewell, A.C.; Haskins, M.E.; Giger, U. Inherited metabolic disease in companion animals: Searching for nature’s mistakes. Vet. J. 2007, 174, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Lavely, J.A. Pediatric seizure disorders in dogs and cats. Vet. Clin. N. Am. Small Anim. Pract. 2014, 44, 275–301. [Google Scholar] [CrossRef]
- Berendt, M.; Farquhar, R.G.; Mandigers, P.J.; Pakozdy, A.; Bhatti, S.F.; De Risio, L.; Fischer, A.; Long, S.; Matiasek, K.; Munana, K.; et al. International veterinary epilepsy task force consensus report on epilepsy definition, classification and terminology in companion animals. BMC Vet. Res. 2015, 11, 182. [Google Scholar] [CrossRef] [Green Version]
- De Risio, L.; Bhatti, S.; Munana, K.; Penderis, J.; Stein, V.; Tipold, A.; Berendt, M.; Farqhuar, R.; Fischer, A.; Long, S.; et al. International veterinary epilepsy task force consensus proposal: Diagnostic approach to epilepsy in dogs. BMC Vet. Res. 2015, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Luttgen, P.; Storts, R. Central Nervous system spongiosus of Saluki Dogs. In Proceedings of the American College of Veterinary Internal Medicine Forum, San Diego, CA, USA, 21–24 May 1987. [Google Scholar]
- Summers, B.; Cummings, J.F.; de Lahunta, A. Veterinary Neuropathology; Mosby: St. Louis, MO, USA, 2005. [Google Scholar]
- Kanekar, S.; Gustas, C. Metabolic disorders of the brain: Part I. Semin Ultrasound CT MR 2011, 32, 590–614. [Google Scholar] [CrossRef]
- Markovich, J.E.; Heinze, C.R.; Freeman, L.M. Thiamine deficiency in dogs and cats. J. Am. Vet. Med. Assoc. 2013, 243, 649–656. [Google Scholar] [CrossRef]
- Brauer, C.; Jambroszyk, M.; Tipold, A. Metabolic and toxic causes of canine seizure disorders: A retrospective study of 96 cases. Vet. J. 2011, 187, 272–275. [Google Scholar] [CrossRef]
- Peterson, M.E. Bromethalin. Top Companion Anim. Med. 2013, 28, 21–23. [Google Scholar] [CrossRef]
- Tauro, A.; Beltran, E.; Cherubini, G.B.; Coelho, A.T.; Wessmann, A.; Driver, C.J.; Rusbridge, C.J. Metronidazole-induced neurotoxicity in 26 dogs. Aust. Vet. J. 2018, 96, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK(1.7): A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Kierczak, M.; Jablonska, J.; Forsberg, S.K.; Bianchi, M.; Tengvall, K.; Pettersson, M.; Scholz, V.; Meadows, J.R.; Jern, P.; Carlborg, O.; et al. Cgmisc: Enhanced genome-wide association analyses and visualization. Bioinformatics 2015, 31, 3830–3831. [Google Scholar] [CrossRef]
- Wickham. Elegant Graphics for Data Analysis, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Mansour, T.A.; Lucot, K.; Konopelski, S.E.; Dickinson, P.J.; Sturges, B.K.; Vernau, K.L.; Choi, S.; Stern, J.A.; Thomasy, S.M.; Doring, S.; et al. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLoS Genet. 2018, 14, e1007850. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Minor, K.M.; Letko, A.; Becker, D.; Drogemuller, M.; Mandigers, P.J.J.; Bellekom, S.R.; Leegwater, P.A.J.; Stassen, Q.E.M.; Putschbach, K.; Fischer, A.; et al. Canine NAPEPLD-associated models of human myelin disorders. Sci. Rep. 2018, 8, 5818. [Google Scholar] [CrossRef]
- Rozen, S.; Skaletsky, H. Methods in Molecular Biology: Bioinformatics Methods and Protocols; Primer3 on the WWW for general users and for biologist programmers; Humana Press: Totowa, NJ, USA, 2000; Volume 132. [Google Scholar]
- Brinkhof, B.; Spee, B.; Rothuizen, J.; Penning, L.C. Development and evaluation of canine reference genes for accurate quantification of gene expression. Anal. Biochem. 2006, 356, 36–43. [Google Scholar] [CrossRef]
- Gibson, K.M.; Aramaki, S.; Sweetman, L.; Nyhan, W.L.; DeVivo, D.C.; Hodson, A.K.; Jakobs, C. Stable isotope dilution analysis of 4-hydroxybutyric acid: An accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. BioMed. Environ. Mass Spectrom. 1990, 19, 89–93. [Google Scholar] [CrossRef]
- Struys, E.A.; Jansen, E.E.; Gibson, K.M.; Jakobs, C. Determination of the GABA analogue succinic semialdehyde in urine and cerebrospinal fluid by dinitrophenylhydrazine derivatization and liquid chromatography-tandem mass spectrometry: Application to SSADH deficiency. J. Inherit. Metab. Dis. 2005, 28, 913–920. [Google Scholar] [CrossRef]
- Gibson, K.M.; Lee, C.F.; Chambliss, K.L.; Kamali, V.; Francois, B.; Jaeken, J.; Jakobs, C. 4-Hydroxybutyric aciduria: Application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin. Chim. Acta 1991, 196, 219–221. [Google Scholar] [CrossRef]
- Pearl, P.L.; Parviz, M.; Vogel, K.; Schreiber, J.; Theodore, W.H.; Gibson, K.M. Inherited disorders of gamma-aminobutyric acid metabolism and advances in ALDH5A1 mutation identification. Dev. Med. Child Neurol. 2015, 57, 611–617. [Google Scholar] [CrossRef]
- Kim, K.J.; Pearl, P.L.; Jensen, K.; Snead, O.C.; Malaspina, P.; Jakobs, C.; Gibson, K.M. Succinic semialdehyde dehydrogenase: Biochemical-molecular-clinical disease mechanisms, redox regulation, and functional significance. Antioxid. Redox Signal. 2011, 15, 691–718. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Kong, X.D.; Kan, Q.C.; Shi, H.R.; Wu, Q.H.; Zhuo, Z.H.; Bai, Q.L.; Jiang, M. Mutation analysis and prenatal diagnosis in a Chinese family with succinic semialdehyde dehydrogenase and a systematic review of the literature of reported ALDH5A1 mutations. J. Perinat. Med. 2016, 44, 441–451. [Google Scholar] [CrossRef]
- Parviz, M.; Vogel, K.; Gibson, K.M.; Pearl, P.L. Disorders of GABA metabolism: SSADH and GABA-transaminase deficiencies. J. Pediatr. Epilepsy 2014, 3, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Jakobs, C.; Bojasch, M.; Mönch, E.; Rating, D.; Siemes, H.; Hanefeld, F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin. Chim. Acta 1981, 111, 169–178. [Google Scholar] [CrossRef]
- DiBacco, M.L.; Roullet, J.B.; Kapur, K.; Brown, M.N.; Walters, D.C.; Gibson, K.M.; Pearl, P.L. Age-related phenotype and biomarker changes in SSADH deficiency. Ann. Clin. Transl. Neurol. 2019, 6, 114–120. [Google Scholar] [CrossRef]
- Pearl, P.L.; Gibson, K.M.; Acosta, M.T.; Vezina, L.G.; Theodore, W.H.; Rogawski, M.A.; Novotny, E.J.; Gropman, A.; Conry, J.A.; Berry, G.T.; et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003, 60, 1413–1417. [Google Scholar] [CrossRef]
- Gibson, K.M.; Christensen, E.; Jakobs, C.; Fowler, B.; Clarke, M.A.; Hammersen, G.; Raab, K.; Kobori, J.; Moosa, A.; Vollmer, B.; et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): Case reports of 23 new patients. Pediatrics 1997, 99, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Knerr, I.; Gibson, K.M.; Murdoch, G.; Salomons, G.S.; Jakobs, C.; Combs, S.; Pearl, P.L. Neuropathology in succinic semialdehyde dehydrogenase deficiency. Pediatr. Neurol. 2010, 42, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Malaspina, P.; Roullet, J.B.; Pearl, P.L.; Ainslie, G.R.; Vogel, K.R.; Gibson, K.M. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem. Int. 2016, 99, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Sgaravatti, A.M.; Sgarbi, M.B.; Testa, C.G.; Durigon, K.; Pederzolli, C.D.; Prestes, C.C.; Wyse, A.T.; Wannmacher, C.M.; Wajner, M.; Dutra-Filho, C.S. Gamma-hydroxybutyric acid induces oxidative stress in cerebral cortex of young rats. Neurochem. Int. 2007, 50, 564–570. [Google Scholar] [CrossRef]
- Struys, E.A.; Verhoeven, N.M.; Jansen, E.E.; Ten Brink, H.J.; Gupta, M.; Burlingame, T.G.; Quang, L.S.; Maher, T.; Rinaldo, P.; Snead, O.C.; et al. Metabolism of gamma-hydroxybutyrate to d-2-hydroxyglutarate in mammals: Further evidence for d-2-hydroxyglutarate transhydrogenase. Metabolism 2006, 55, 353–358. [Google Scholar] [CrossRef]
- Teter, C.J.; Guthrie, S.K. A comprehensive review of MDMA and GHB: Two common club drugs. Pharmacotherapy 2001, 21, 1486–1513. [Google Scholar] [CrossRef] [Green Version]
- Kamal, R.M.; van Noorden, M.S.; Franzek, E.; Dijkstra, B.A.; Loonen, A.J.; De Jong, C.A. The Neurobiological Mechanisms of Gamma-Hydroxybutyrate Dependence and Withdrawal and Their Clinical Relevance: A Review. Neuropsychobiology 2016, 73, 65–80. [Google Scholar] [CrossRef]
- Morris, M.E.; Hu, K.; Wang, Q. Renal clearance of gamma-hydroxybutyric acid in rats: Increasing renal elimination as a detoxification strategy. J. Pharmacol. Exp. Ther. 2005, 313, 1194–1202. [Google Scholar] [CrossRef] [Green Version]
- Felmlee, M.A.; Wang, Q.; Cui, D.; Roiko, S.A.; Morris, M.E. Mechanistic toxicokinetic model for gamma-hydroxybutyric acid: Inhibition of active renal reabsorption as a potential therapeutic strategy. AAPS J. 2010, 12, 407–416. [Google Scholar] [CrossRef]
- Shinka, T.; Ohfu, M.; Hirose, S.; Kuhara, T. Effect of valproic acid on the urinary metabolic profile of a patient with succinic semialdehyde dehydrogenase deficiency. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 792, 99–106. [Google Scholar] [CrossRef]
- Didiasova, M.; Banning, A.; Brennenstuhl, H.; Jung-Klawitter, S.; Cinquemani, C.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: An Update. Cells 2020, 9, 477. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.R.; Ainslie, G.R.; Walters, D.C.; McConnell, A.; Dhamne, S.C.; Rotenberg, A.; Roullet, J.B.; Gibson, K.M. Succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism: An update on pharmacological and enzyme-replacement therapeutic strategies. J. Inherit. Metab. Dis. 2018, 41, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Drasbek, K.R.; Vardya, I.; Delenclos, M.; Gibson, K.M.; Jensen, K. SSADH deficiency leads to elevated extracellular GABA levels and increased GABAergic neurotransmission in the mouse cerebral cortex. J. Inherit. Metab. Dis. 2008, 31, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Gibson, K.M.; Jakobs, C.; Pearl, P.L.; Snead, O.C. Murine succinate semialdehyde dehydrogenase (SSADH) deficiency, a heritable disorder of GABA metabolism with epileptic phenotype. IUBMB Life 2005, 57, 639–644. [Google Scholar] [CrossRef]
- Gupta, M.; Hogema, B.M.; Grompe, M.; Bottiglieri, T.G.; Concas, A.; Biggio, G.; Sogliano, C.; Rigamonti, A.E.; Pearl, P.L.; Snead, O.C., 3rd; et al. Murine succinate semialdehyde dehydrogenase deficiency. Ann. Neurol. 2003, 54 (Suppl. 6), S81–S90. [Google Scholar] [CrossRef]
- Kelmer, E.; Gibson, K.M.; Jakobs, C.; Struys, E.; Shelton, G.D.; Aroch, I.; O’Brien, D.P. Severe Lactic Acidosis Associated with a Suspected Succinic Semialdehyde Dehydrogenas (SSADH) Deficiency in a Young Chihuahua Dog. Isr. J. Vet. Med. 2018, 73, 43–48. [Google Scholar]
- Pearl, P.L.; Wiwattanadittakul, N.; Roullet, J.B.; Gibson, K.M. Succinic Semialdehyde Dehydrogenase Deficiency. In GeneReviews (R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pearl, P.L.; Gibson, K.M.; Cortez, M.A.; Wu, Y.; Carter Snead, O., 3rd; Knerr, I.; Forester, K.; Pettiford, J.M.; Jakobs, C.; Theodore, W.H. Succinic semialdehyde dehydrogenase deficiency: Lessons from mice and men. J. Inherit. Metab. Dis. 2009, 32, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Lu, Y.; Morris, M.E. Monocarboxylate transporter (MCT) mediates the transport of gamma-hydroxybutyrate in human kidney HK-2 cells. Pharm. Res. 2007, 24, 1067–1078. [Google Scholar] [CrossRef]
- Busardo, F.P.; Zaami, S.; Baglio, G.; Indorato, F.; Montana, A.; Giarratana, N.; Kyriakou, C.; Marinelli, E.; Romano, G. Assessment of the stability of exogenous gamma hydroxybutyric acid (GHB) in stored blood and urine specimens. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4187–4194. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dog No. | Sample ID | Country | Sex | Age of Onset | Clinical Signs | Neurological Examination | Outcome |
|---|---|---|---|---|---|---|---|
| 1 | 5813 | USA | F | 10 weeks | Generalized epileptic seizures, episodes of vocalization, abnormal behavior, generalized ataxia with thoracic limb hypermetria | Mild generalized ataxia with thoracic limb hypermetria. Delayed proprioceptive positioning present in all 4 limbs | Treated with anticonvulsants (phenobarbital), euthanized at 32 weeks of age |
| 2 | 4942 | USA | F | 10 weeks | Generalized epileptic seizures, episodes of vocalization, abnormal behavior, generalized ataxia with thoracic limb hypermetria | Not done | Treated with anticonvulsants (phenobarbital), euthanized at 39 weeks of age |
| 3 | 1053 | USA | F | 6 weeks | Focal epileptic seizures, episodes of vocalization, normal between episodes Unable to arouse when sleeping | Absent menace response OU | Treated with anticonvulsants (phenobarbital), euthanized at 17 weeks of age |
| 4 | 1052 | USA | M | 6 weeks | Generalized and focal epileptic seizures, episodes of vocalization, normal between episodes. Unable to arouse when sleeping | Absent menace response OU | Treated with anticonvulsants (phenobarbital), euthanized at 17 weeks of age |
| 5 | SL006 | Germany | M | 9 weeks | Focal epileptic seizures, deep sleep | Thoracic limb hypermetria, mild ataxia, reduced proprioceptive positioning, absent menace | Treated with levetiracetam, euthanized at 4 months of age |
| 6 | SL008 | Germany | M | 9 weeks | Focal epileptic seizures, episodes of vocalization | Not done | Euthanized at unknown age |
| 7 | SL007 | Germany | F | 9 weeks | Focal epileptic seizures | Not done | Euthanized at unknown age |
| Dog Number | Urine SSA, mmol/mol Creatinine | Urine GHB, mmol/mol Creatinine | Urine DHHA mmol/mol Creatinine | Serum GHB, µmol/L | Serum DHHA, µmol/L | CSF SSA, µmol/L | CSF GHB, µmol/L | CSF DHHA, µmol/L | Brain GHB, nmol/mg Brain | Brain DHHA, nmol/mg Brain | Brain SSA, nmol/mg Brain | Brain SSA Activity, pmol/min/mg Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9.23 | 1.06 | 5.85 | 6.59 | 0.45 | 69 | >1500 | 43.3 | 2.43 | 0.22 | 0.23 | 10 |
| 2 | nd | nd | nd | nd | nd | nd | nd | nd | 2.93 | 0.28 | 0.18 | 0 |
| 5 | 38.7 | nd | 11.8 | nd | 0.41 | nd | nd | nd | nd | nd | nd | nd |
| 6 | 30.9 | 0.67 | 10.4 | nd | 0.61 | nd | nd | nd | nd | nd | nd | nd |
| 7 | nd | nd | nd | nd | 0.56 | nd | nd | nd | nd | nd | nd | nd |
| Number of affected dogs | 3 | 2 | 3 | 1 | 4 | 1 | 1 | 1 | 2 | nd | 2 | 2 |
| Number of control dogs | 4 | 4 | 4 | 4 | 4 | 2 | 3 | 3 | 4 | nd | nd | 4 |
| median affected | 30.9 | 0.87 | 10.4 | n/a | 0.51 | n/a | n/a | n/a | 2.68 | 0.25 | 0.21 | 5 |
| range affected | 9.23–38.7 | 0.67–1.06 | 5.85–11.8 | n/a | 0.41–0.61 | n/a | n/a | n/a | 2.43–2.93 | 0.22–0.28 | 0.18–0.23 | 0–10 |
| median control | 0.86 | 0.82 | 0.29 | 0.38 | 0.08 | 0.24 | 0.31 | 0.11 | 0.03 | 0 | 0.11 | 5587 |
| range control | 0.64–0.9 | 0.29–2.04 | 0.18–0.65 | 0.28–0.59 | 0.07–0.1 | 0.02–0.46 | 0.23–0.8 | 0.1–0.2 | 0–0.05 | 0–0 | 0.06–0.14 | 4214–5942 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernau, K.M.; Struys, E.; Letko, A.; Woolard, K.D.; Aguilar, M.; Brown, E.A.; Cissell, D.D.; Dickinson, P.J.; Shelton, G.D.; Broome, M.R.; et al. A Missense Variant in ALDH5A1 Associated with Canine Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD) in the Saluki Dog. Genes 2020, 11, 1033. https://doi.org/10.3390/genes11091033
Vernau KM, Struys E, Letko A, Woolard KD, Aguilar M, Brown EA, Cissell DD, Dickinson PJ, Shelton GD, Broome MR, et al. A Missense Variant in ALDH5A1 Associated with Canine Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD) in the Saluki Dog. Genes. 2020; 11(9):1033. https://doi.org/10.3390/genes11091033
Chicago/Turabian StyleVernau, Karen M., Eduard Struys, Anna Letko, Kevin D. Woolard, Miriam Aguilar, Emily A. Brown, Derek D. Cissell, Peter J. Dickinson, G. Diane Shelton, Michael R. Broome, and et al. 2020. "A Missense Variant in ALDH5A1 Associated with Canine Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD) in the Saluki Dog" Genes 11, no. 9: 1033. https://doi.org/10.3390/genes11091033
APA StyleVernau, K. M., Struys, E., Letko, A., Woolard, K. D., Aguilar, M., Brown, E. A., Cissell, D. D., Dickinson, P. J., Shelton, G. D., Broome, M. R., Gibson, K. M., Pearl, P. L., König, F., Van Winkle, T. J., O’Brien, D., Roos, B., Matiasek, K., Jagannathan, V., Drögemüller, C., ... Bannasch, D. L. (2020). A Missense Variant in ALDH5A1 Associated with Canine Succinic Semialdehyde Dehydrogenase Deficiency (SSADHD) in the Saluki Dog. Genes, 11(9), 1033. https://doi.org/10.3390/genes11091033