LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Selection
2.3. Histopathological Examinations
2.4. Whole Genome Sequencing
2.5. Variant Calling
2.6. Gene Analysis
2.7. Sanger Sequencing
3. Results
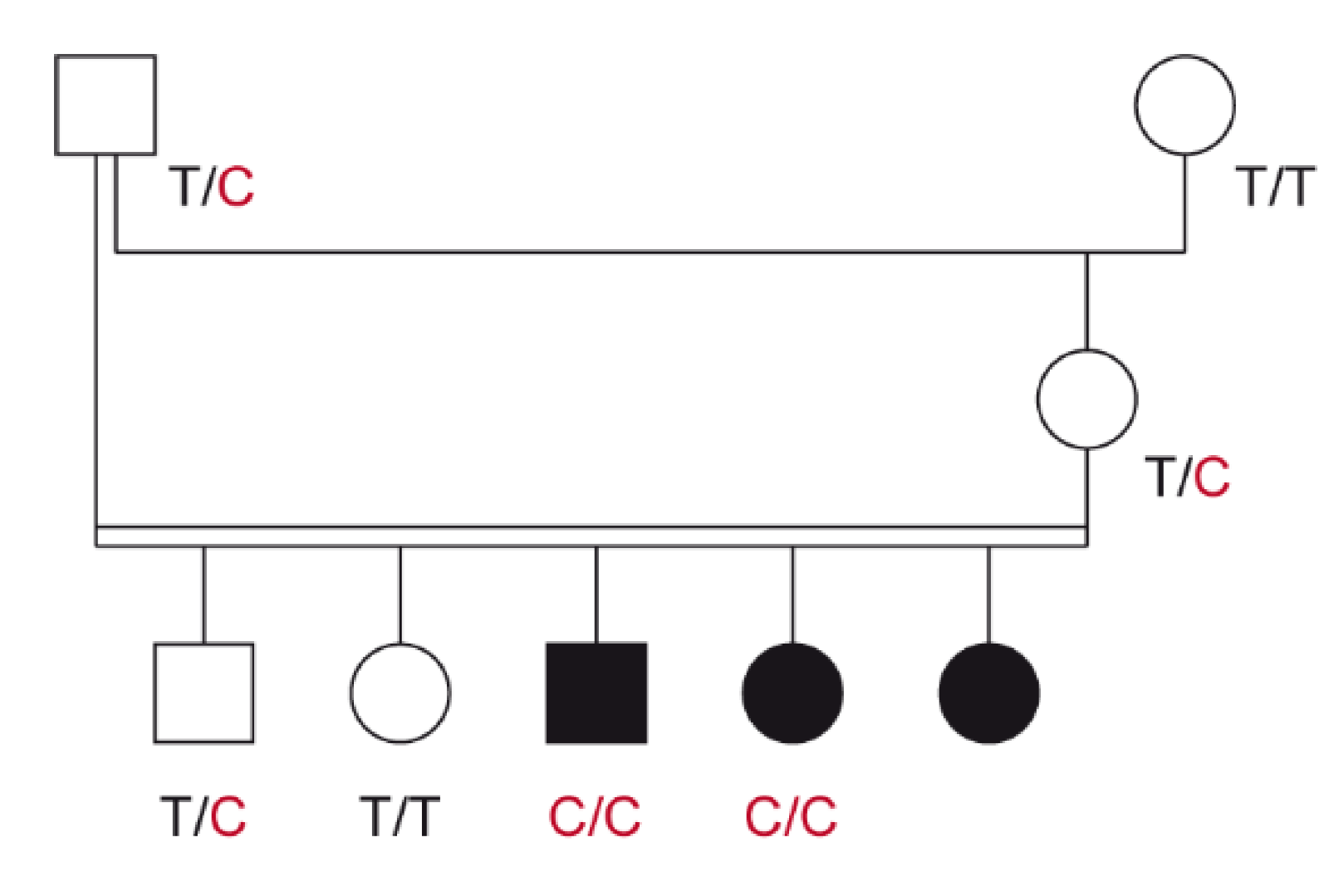
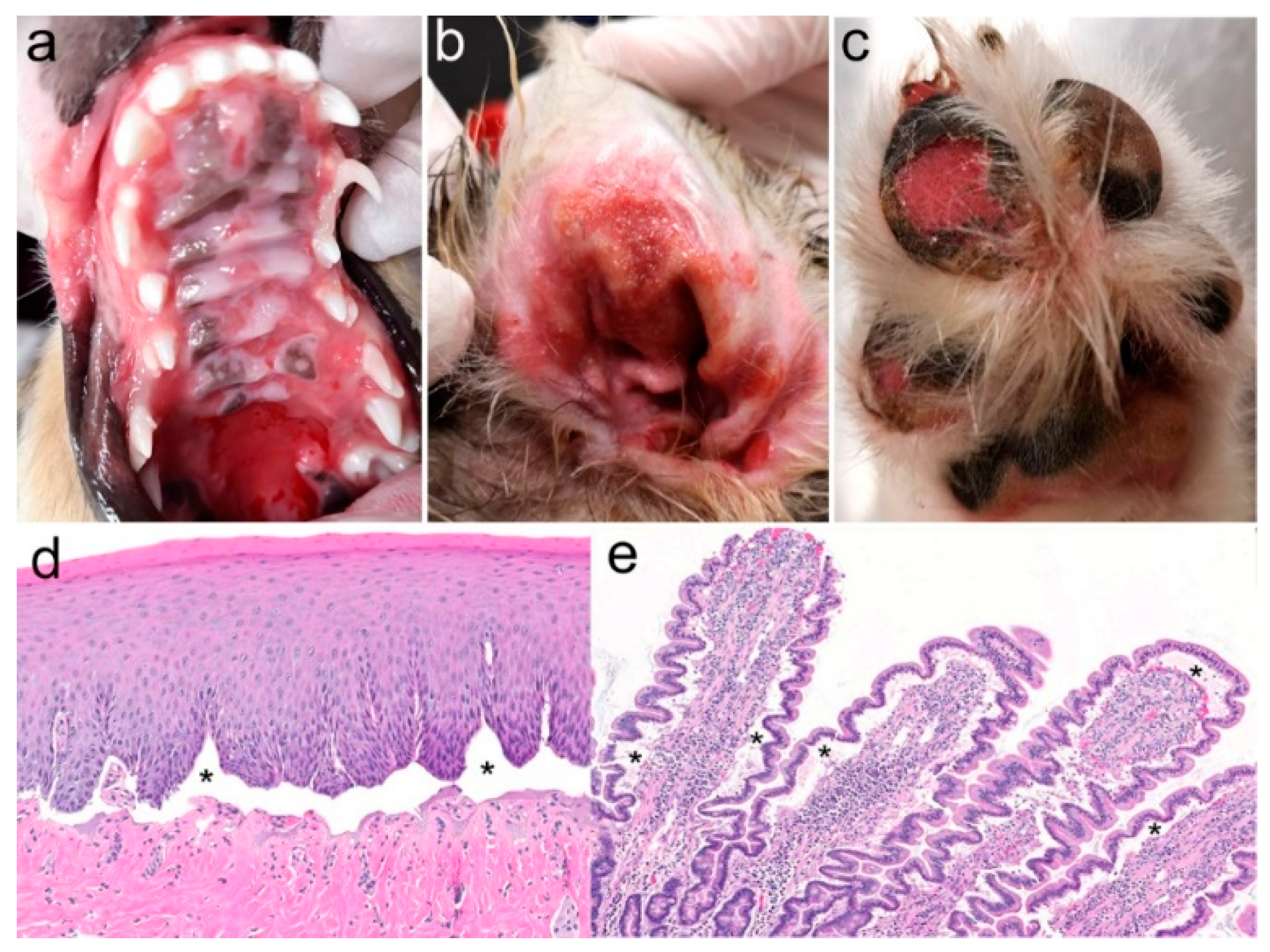
3.1. Family Anamnesis, Clinical Examinations, and Histopathology
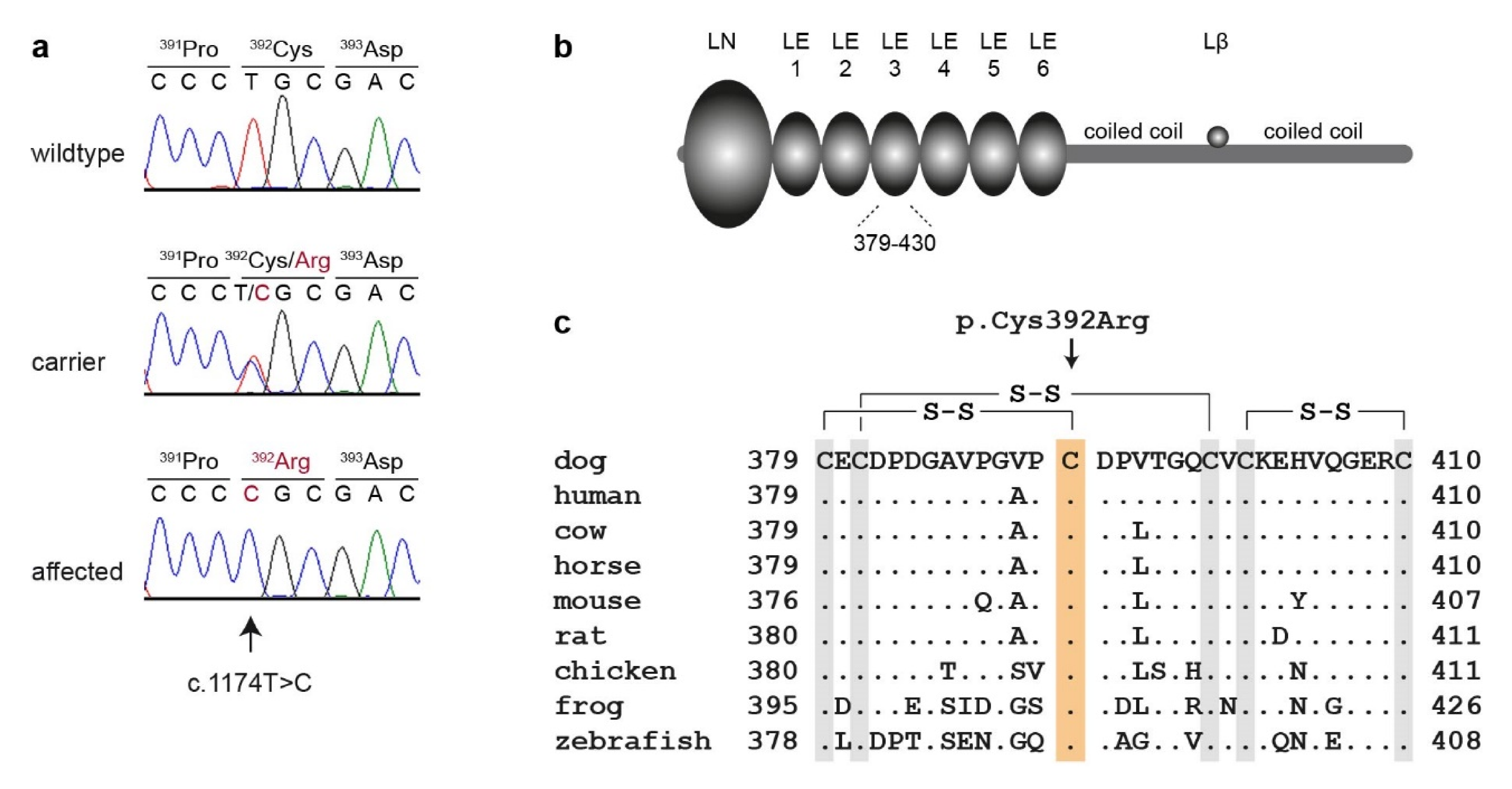
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Credille, K.M.; Barnhart, K.F.; Minor, J.S.; Dunstan, R.W. Mild recessive epidermolytic hyperkeratosis associated with a novel keratin 10 donor splice-site mutation in a family of Norfolk terrier dogs. Br. J. Dermatol. 2005, 153, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivry, T.; Linder, K.E.; Wang, P.; Bizikova, P.; Bernstein, J.A.; Dunston, S.M.; Paps, J.S.; Casal, M.L. Deficient plakophilin-1 expression due to a mutation in PKP1 causes ectodermal dysplasia-skin fragility syndrome in Chesapeake Bay Retriever dogs. PLoS ONE 2012, 7, e32072. [Google Scholar] [CrossRef] [Green Version]
- Mauldin, E.A.; Wang, P.; Olivry, T.; Henthorn, P.S.; Casal, M.L. Epidermolysis bullosa simplex in sibling Eurasier dogs is caused by a PLEC non-sense variant. Vet. Dermatol. 2017, 28, 10-e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capt, A.; Spirito, F.; Guaguere, E.; Spadafora, A.; Ortonne, J.P.; Meneguzzi, G. Molecular basis of inherited junctional epidermolysis bullosa in the German pointer: Establishment of a large animal model for the condition. J. Investig. Dermatol. 2005, 124, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldeschi, C.; Gache, Y.; Rattenholl, A.; Bouille, P.; Danos, O.; Ortonne, J.P.; Bruckner-Tuderman, L.; Meneguzzi, G. Genetic correction of canine dystrophic epidermolysis bullosa mediated by retroviral vectors. Hum. Mol. Genet. 2003, 12, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niskanen, J.; Dillard, K.; Arumilli, M.; Salmela, E.; Anttila, M.; Lohi, H.; Hytönen, M.K. Nonsense variant in COL7A1 causes recessive dystrophic epidermolysis bullosa in Central Asian Shepherd dogs. PLoS ONE 2017, 12, e0177527. [Google Scholar] [CrossRef]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–553. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Has, C.; Nystrom, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71, 313–329. [Google Scholar] [CrossRef]
- Chen, M.; Marinkovich, M.P.; Jones, J.C.; O’Toole, E.A.; Li, Y.Y.; Woodley, D.T. NC1 domain of type VII collagen binds to the beta3 chain of laminin 5 via a unique subdomain within the fibronectin-like repeats. J. Investig. Dermatol. 1999, 112, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, K.T.; Henney, P.J.; Ennis, R.B. Partial deletion of the LAMA3 gene is responsible for hereditary junctional epidermolysis bullosa in the American Saddlebred Horse. Anim. Genet. 2009, 40, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Spirito, F.; Charlesworth, A.; Linder, K.; Ortonne, J.P.; Baird, J.; Meneguzzi, G. Animal models for skin blistering conditions: Absence of laminin 5 causes hereditary junctional mechanobullous disease in the Belgian horse. J. Investig. Dermatol. 2002, 119, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, D.; Chaffaux, S.; Taourit, S.; Guerin, G. A mutation in the LAMC2 gene causes the Herlitz junctional epidermolysis bullosa (H-JEB) in two French draft horse breeds. Genet. Sel. Evol. 2003, 35, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, K.; Brachelente, C.; Passamonti, F.; Flati, A.; Silvestrelli, M.; Capomaccio, S. First report of junctional epidermolysis bullosa (JEB) in the Italian draft horse. BMC Vet. Res. 2015, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, A.; Chao, S.C.; Pulkkinen, L.; Murrell, D.; Bruckner-Tuderman, L.; Pfendner, E.; Uitto, J. Laminin 5 mutations in junctional epidermolysis bullosa: Molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum. Genet. 2002, 110, 41–51. [Google Scholar] [CrossRef]
- Varki, R.; Sadowski, S.; Pfendner, E.; Uitto, J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J. Med. Genet. 2006, 43, 641–652. [Google Scholar] [CrossRef]
- Kiritsi, D.; Has, C.; Bruckner-Tuderman, L. Laminin 332 in junctional epidermolysis bullosa. Cell Adh. Migr. 2013, 7, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Alhaidari, Z.; Olivry, T.; Spadafora, A.; Thomas, R.C.; Perrin, C.; Meneguzzi, G.; Ortonne, J.P. Junctional epidermolysis bullosa in two domestic shorthaired kittens. Vet. Dermatol. 2005, 16, 69–73. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Website, Entry Q13751. Available online: https://www.uniprot.org/uniprot/Q13751 (accessed on 20 May 2020).
- Olivry, T.; Poujade-Delverdier, A.; Dunston, S.M.; Fine, J.-D.; Ortonne, J.-P. Absent expression of collagen XVII (BPAG2, BP180) in canine familial localized junctional epidermolysis bullosa. Vet. Dermatol. 1997, 8, 203–212. [Google Scholar] [CrossRef]
- Guaguère, E.; Olivry, T.; Poujade-Delverdier, A.; Magnol, J.P. Epidermolyse bulleuse jonctionnelle familiale associée à une absence d’expression de collagène XVII chez le Braque Allemand: À propos de deux cas. Prat. Méd. Chir. Anim. Comp. 1997, 32, 471–480. [Google Scholar]
- GeneCards: The Human Gene Database. Available online: https://www.genecards.org (accessed on 10 August 2020).
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Engel, J. EGF-like domains in extracellular matrix proteins: Localized signals for growth and differentiation? FEBS Lett. 1989, 251, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Beck, K.; Hunter, I.; Engel, J. Structure and function of laminin: Anatomy of a multidomain glycoprotein. FASEB J. 1990, 4, 148–160. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disorder | Level of Cleavage | Gene | Protein | Inheritance 1 |
|---|---|---|---|---|
| Classical Epidermolysis Bullosa (EB) | ||||
| EB simplex (EBS) | Basal epidermal | CD151 | CD151 molecule (Raph blood group) | AR |
| DST | dystonin | AR | ||
| EXPH5 | exophilin 5 | AR | ||
| KLHL24 | kelch like family member 24 | AD | ||
| KRT5 | keratin 5 | AD, AR | ||
| KRT14 | keratin 14 | AD, AR | ||
| PLEC | plectin | AR | ||
| Junctional EB (JEB) | Junctional | COL17A1 | collagen type XVII, α 1 chain | AR |
| ITGA3 | integrin subunit α 3 | AR | ||
| ITGA6 | integrin subunit α 6 | AR | ||
| ITGB4 | integrin subunit β 4 | AR | ||
| LAMA3 | laminin subunit α 3 | AR | ||
| LAMB3 | laminin subunit β 3 | AR | ||
| LAMC2 | laminin subunit γ 2 | AR | ||
| Dystrophic EB (DEB) | Dermal | COL7A1 | collagen type VII, α 1 chain | AD, AR |
| Kindler EB | Mixed | FERMT1 | fermitin family homolog 1 | AR |
| Other Disorders with Skin Fragility | ||||
| Peeling skin disorders | Intraepidermal | CAST | calpastatin | AR |
| CSTA | cystatin A | AR | ||
| CTSB | cystatin B | AR | ||
| DSG1 | desmoglein 1 | AR | ||
| FLG2 | filaggrin family member 2 | AR | ||
| SERPINB8 | serpin family B member 8 | AR | ||
| SPINK5 | serine peptidase inhibitor Kazal type 5 | AR | ||
| Erosive skin fragility disorders | Intraepidermal | DSC3 | desmocollin 3 | AR |
| DSG3 | desmoglein 3 | AR | ||
| DSP | desmoplakin | AR | ||
| JUP | junction plakoglobin | AR | ||
| PKP1 | plakophilin 1 | AR | ||
| Keratinopathic ichthyoses | Intraepidermal | KRT1 | keratin 1 | AD |
| KRT2 | keratin 2 | AD | ||
| KRT10 | keratin 10 | AD, AR | ||
| Pachyonychia congenita | Intraepidermal | KRT6A | keratin 6A | AD |
| KRT6B | keratin 6B | AD | ||
| KRT6C | keratin 6C | AD | ||
| KRT16 | keratin 16 | AD | ||
| KRT17 | keratin 17 | AD | ||
| Syndromic connective tissue disorder with skin fragility | Dermal | PLOD3 | procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | AR |
| Filtering Step | Variants |
|---|---|
| All variants in the affected dog | 3,111,811 |
| Private variants | 11,754 |
| Protein-changing private variants | 54 |
| Protein-changing private variants in 37 candidate genes | 1 |
| Dogs | T/T | T/C | C/C |
|---|---|---|---|
| Cases (n = 2) 1 | - | - | 2 |
| Controls, Australian Shepherd dogs (n = 245) | 242 | 3 | - |
| Controls, other breeds (n = 663) 1 | 663 | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiener, S.; Laprais, A.; Mauldin, E.A.; Jagannathan, V.; Olivry, T.; Leeb, T. LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa. Genes 2020, 11, 1055. https://doi.org/10.3390/genes11091055
Kiener S, Laprais A, Mauldin EA, Jagannathan V, Olivry T, Leeb T. LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa. Genes. 2020; 11(9):1055. https://doi.org/10.3390/genes11091055
Chicago/Turabian StyleKiener, Sarah, Aurore Laprais, Elizabeth A. Mauldin, Vidhya Jagannathan, Thierry Olivry, and Tosso Leeb. 2020. "LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa" Genes 11, no. 9: 1055. https://doi.org/10.3390/genes11091055
APA StyleKiener, S., Laprais, A., Mauldin, E. A., Jagannathan, V., Olivry, T., & Leeb, T. (2020). LAMB3 Missense Variant in Australian Shepherd Dogs with Junctional Epidermolysis Bullosa. Genes, 11(9), 1055. https://doi.org/10.3390/genes11091055