LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Myogenic Differentiation
2.2. Morphological Features Determination
2.3. Plasmids and Mutagenesis
2.4. Lentivirus Production, Infection, and Establishment of Stable Cell Lines
2.5. Library Construction and RNA-Seq
2.6. Relative Quantification of mtDNA Copy Number Using Real-Time PCR
2.7. Cell Respiration
2.8. Glycolysis Stress Assay
2.9. Cell Proliferation Assay
2.10. Western Blotting
2.11. Immunocytochemistry
2.12. MitoTracker Staining
2.13. Q-PCR
2.14. Processing of RNA-Seq Data
3. Results
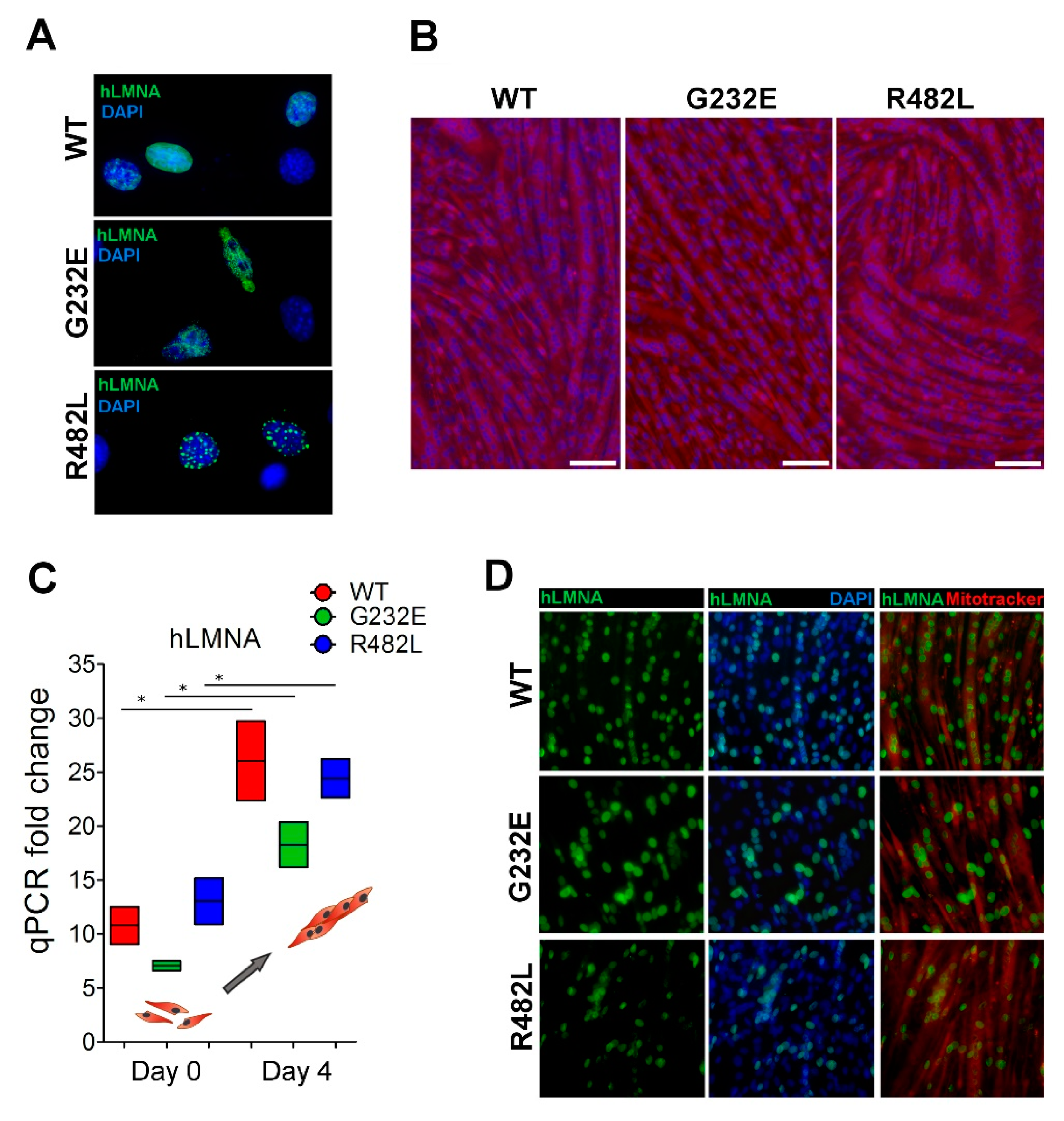
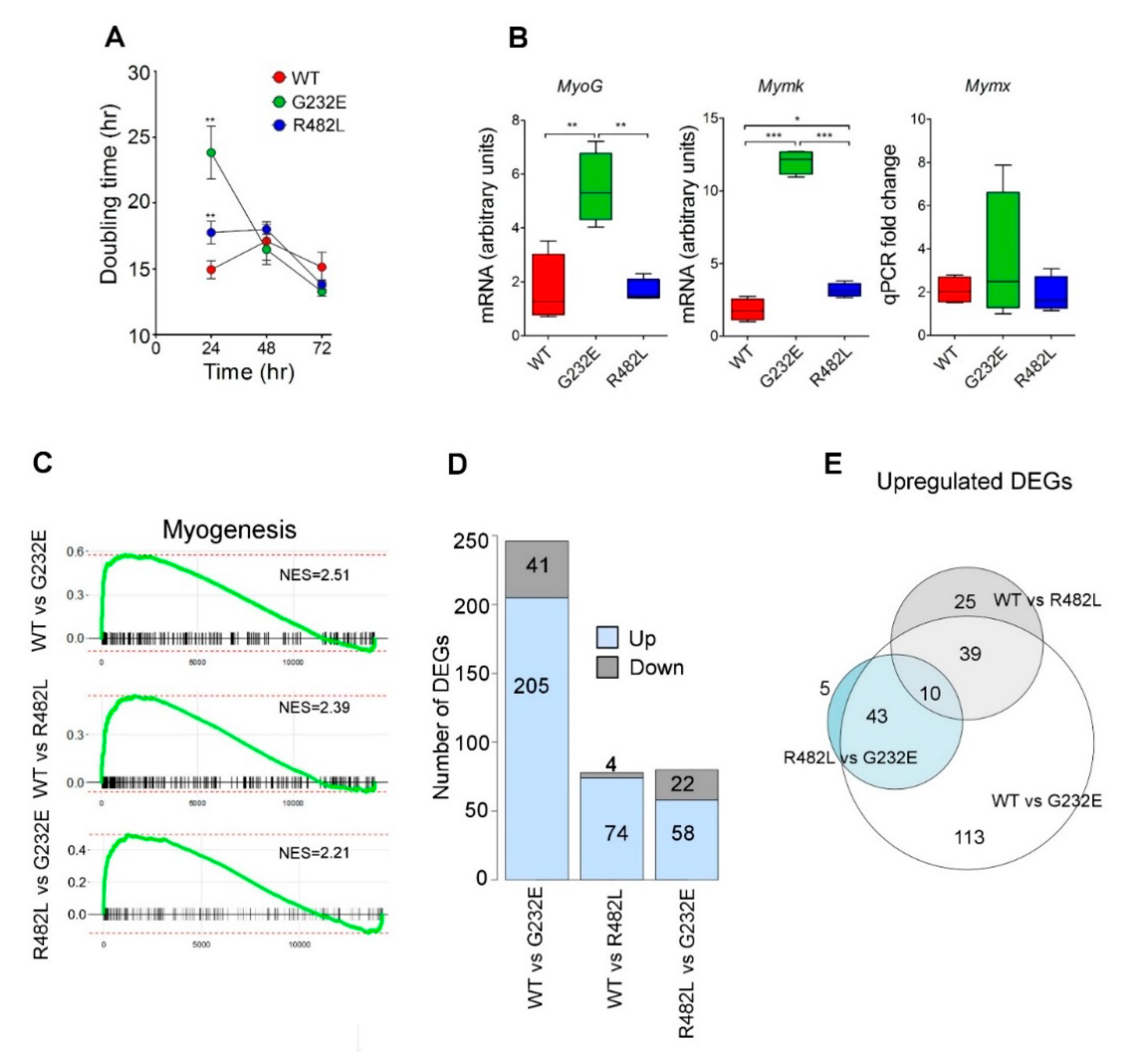
3.1. LMNA Mutations Affect Both Proliferation Activity and Myogenic Commitment in Transgenic C2C12 Myoblasts
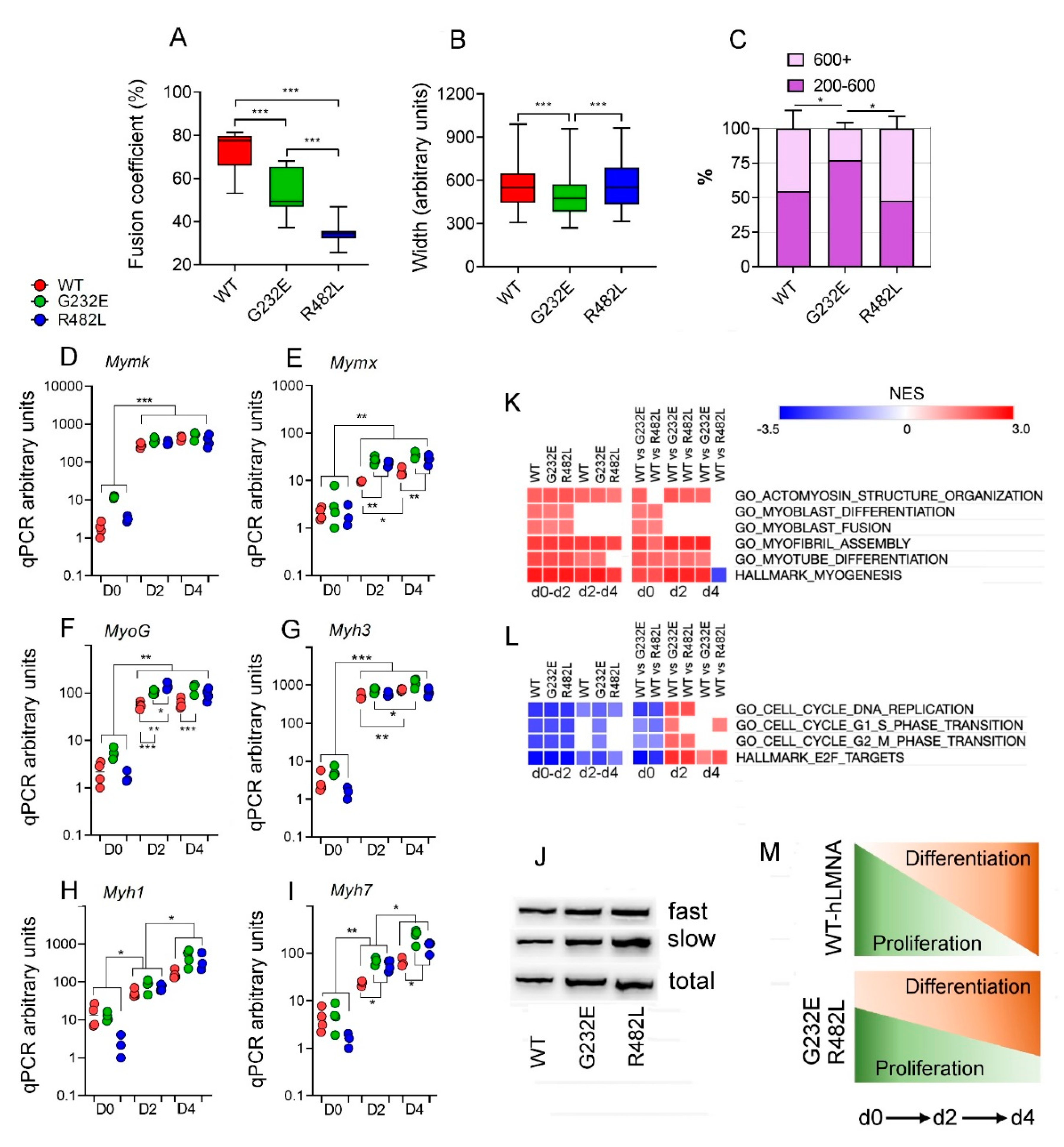
3.2. LMNA Mutations Affect Differentiation Efficacy and Dynamics in Transgenic C2C12 Myoblasts
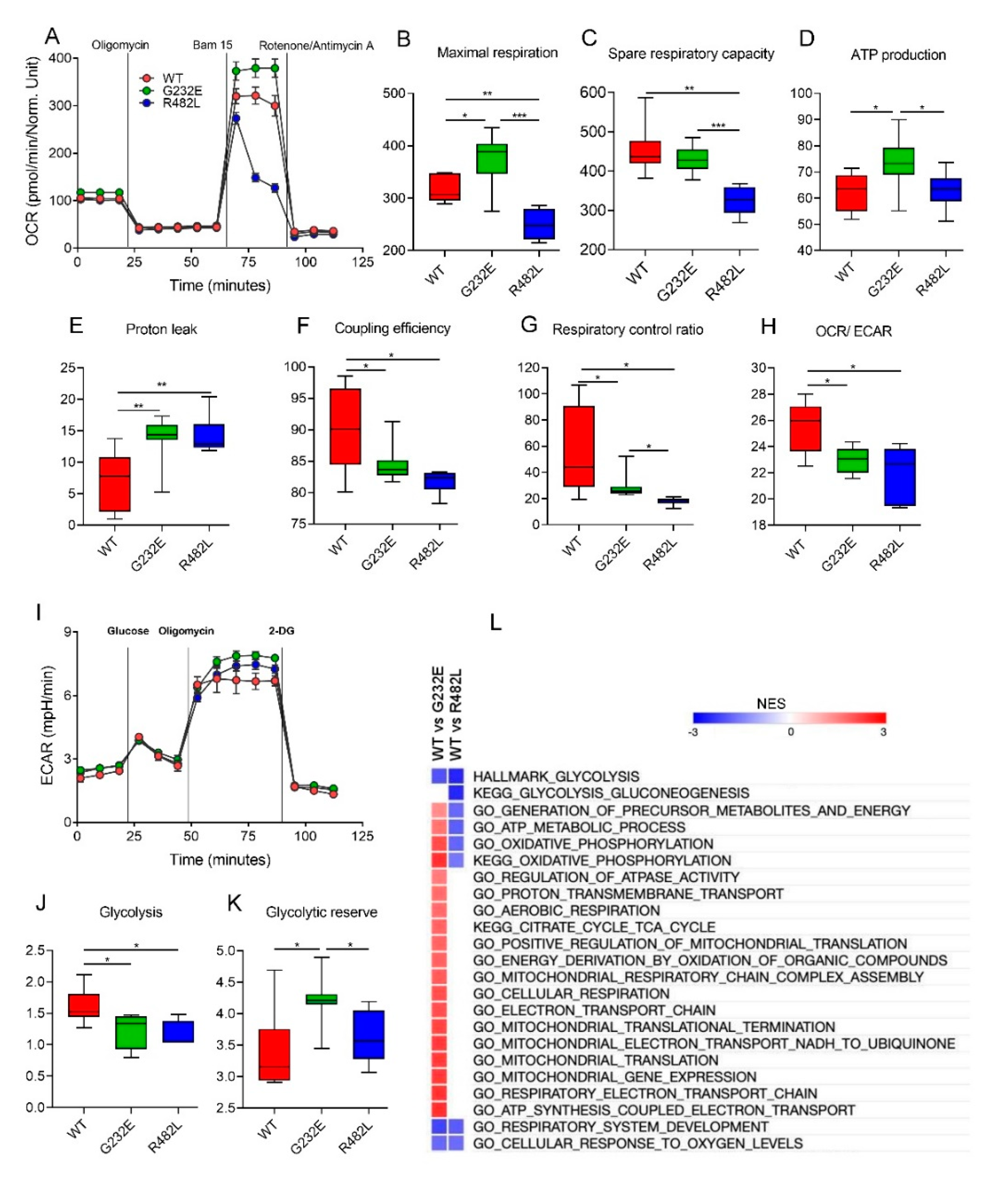
3.3. LMNA Mutations Affect Cellular Bioenergetics of C2C12 Myotubes
4. Discussion
5. Study Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonne, G.; Mercuri, E.; Muchir, A.; Urtizberea, A.; Bécane, H.M.; Recan, D.; Merlini, L.; Wehnert, M.; Boor, R.; Reuner, U.; et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 2000, 48, 170–180. [Google Scholar] [CrossRef]
- Hegele, R.A. LMNA mutation position predicts organ system involvement in laminopathies. Clin. Genet. 2005, 68, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Dobrzynska, A.; Gonzalo, S.; Shanahan, C.; Askjaer, P. The nuclear lamina in health and disease. Nucleus 2016, 7, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigouroux, C.; Guénantin, A.-C.; Vatier, C.; Capel, E.; Le Dour, C.; Afonso, P.; Bidault, G.; Béréziat, V.; Lascols, O.; Capeau, J.; et al. Lipodystrophic syndromes due to LMNA mutations: Recent developments on biomolecular aspects, pathophysiological hypotheses and therapeutic perspectives. Nucleus 2018, 9, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briand, N.; Collas, P. Laminopathy-causing lamin A mutations reconfigure lamina-associated domains and local spatial chromatin conformation. Nucleus 2018, 9, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Rowat, A.C.; Lammerding, J.; Ipsen, J.H. Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophys. J. 2006, 91, 4649–4664. [Google Scholar] [CrossRef] [Green Version]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; Te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Foisner, R. The structural and gene expression hypotheses in laminopathic diseases —Not so different after all. Mol. Biol. Cell 2019, 30, 1786–1790. [Google Scholar] [CrossRef]
- Miroshnikova, Y.A.; Nava, M.M.; Wickström, S.A. Emerging roles of mechanical forces in chromatin regulation. J. Cell Sci. 2017, 130, 2243–2250. [Google Scholar] [CrossRef] [Green Version]
- Gnocchi, V.F.; Ellis, J.A.; Zammit, P.S. Does satellite cell dysfunction contribute to disease progression in Emery-Dreifuss muscular dystrophy? Biochem. Soc. Trans. 2008, 36, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, R.I.; Lelyavina, T.A.; Komarova, M.Y.; Galenko, V.L.; Ivanova, O.A.; Tikanova, P.A.; Khromova, N.V.; Golovkin, A.S.; Bortsova, M.A.; Sergushichev, A.; et al. Skeletal muscle resident progenitor cells coexpress mesenchymal and myogenic markers and are not affected by chronic heart failure-induced dysregulations. Stem Cells Int. 2019, 2019, 5690345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouly, V.; Aamiri, A.; Bigot, A.; Cooper, R.N.; Di Donna, S.; Furling, D.; Gidaro, T.; Jacquemin, V.; Mamchaoui, K.; Negroni, E.; et al. The mitotic clock in skeletal muscle regeneration, disease and cell mediated gene therapy. Acta Physiol. Scand. 2005, 184, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.T.; Worman, H.J. The Nuclear Envelope as a Signaling Node in Development and Disease. Dev. Cell 2009, 17, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Perovanovic, J.; Dell’Orso, S.; Gnochi, V.F.; Jaiswal, J.K.; Sartorelli, V.; Vigouroux, C.; Mamchaoui, K.; Mouly, V.; Bonne, G.; Hoffman, E.P. Laminopathies disrupt epigenomic developmental programs and cell fate. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in Duchenne Muscular Dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.R.; Von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Charar, C.; Gruenbaum, Y. Lamins and metabolism. Clin. Sci. 2017, 131, 105–111. [Google Scholar] [CrossRef]
- Zaremba-Czogalla, M.; Dubińska-Magiera, M.; Rzepecki, R. Laminopathies: The molecular background of the disease and the prospects for its treatment. Cell. Mol. Biol. Lett. 2011, 16, 114–148. [Google Scholar] [CrossRef] [PubMed]
- Boschmann, M.; Engeli, S.; Moro, C.; Luedtke, A.; Adams, F.; Gorzelniak, K.; Rahn, G.; Mähler, A.; Dobberstein, K.; Krüger, A.; et al. LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance. J. Clin. Endocrinol. Metab. 2010, 95, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.; Auclair, M.; Donadille, B.; Béréziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Sieprath, T.; Corne, T.D.J.; Nooteboom, M.; Grootaert, C.; Rajkovic, A.; Buysschaert, B.; Robijns, J.; Broers, J.L.V.; Ramaekers, F.C.S.; Koopman, W.J.H.; et al. Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus 2015, 6, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Weatherall, P.; Adams-Huet, B.; Garg, A. Increased skeletal muscle volume in women with familial partial lipodystrophy, Dunnigan variety. J. Clin. Endocrinol. Metab. 2013, 98, E1410–E1413. [Google Scholar] [CrossRef] [Green Version]
- Khromova, N.V.; Perepelina, K.I.; Ivanova, O.A.; Malashicheva, A.B.; Kostareva, A.A.; Dmitrieva, R.I. R482L Mutation of the LMNA Gene Affects Dynamics of C2C12 Myogenic Differentiation and Stimulates Formation of Intramuscular Lipid Droplets. Biochemistry 2019, 84, 241–249. [Google Scholar] [CrossRef]
- Vantyghem, M.C.; Pigny, P.; Maurage, C.A.; Rouaix-Emery, N.; Stojkovic, T.; Cuisset, J.M.; Millaire, A.; Lascols, O.; Vermersch, P.; Wemeau, J.L.; et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482W mutation show muscular and cardiac abnormalities. J. Clin. Endocrinol. Metab. 2004, 89, 5337–5346. [Google Scholar] [CrossRef] [Green Version]
- Guénantin, A.C.; Briand, N.; Bidault, G.; Afonso, P.; Béréziat, V.; Vatier, C.; Lascols, O.; Caron-Debarle, M.; Capeau, J.; Vigouroux, C. Nuclear envelope-related lipodystrophies. Semin. Cell Dev. Biol. 2014, 29, 148–157. [Google Scholar] [CrossRef]
- Perepelina, K.I.; Smolina, N.A.; Zabirnik, A.S.; Dmitrieva, R.I.; Malashicheva, A.B.; Kostareva, A.A. The role of LMNA mutations in myogenic differentiation of C2C12 and primary satellite cells. Cell Tissue Biol. 2017, 11, 213–219. [Google Scholar] [CrossRef]
- Malashicheva, A.; Kanzler, B.; Tolkunova, E.; Trono, D.; Tomilin, A. Lentivirus as a tool for lineage-specific gene manipulations. Genesis 2007, 45, 456–459. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Rooney, J.P.; Ryde, I.T.; Anbalagan, C.; Joglekar, R.; Meyer, J.N. PCR-Based Analysis of Mitochondrial DNA Copy Number, Mitochondrial DNA Damage, and Nuclear DNA Damage. Curr. Protoc. Toxicol. 2016, 67, 20.11.1–20.11.25. [Google Scholar] [CrossRef]
- Smolina, N.; Khudiakov, A.; Knyazeva, A.; Zlotina, A.; Sukhareva, K.; Kondratov, K.; Gogvadze, V.; Zhivotovsky, B.; Sejersen, T.; Kostareva, A. Desmin mutations result in mitochondrial dysfunction regardless of their aggregation properties. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. Subread manual 1.5.0-p1. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergushichev, A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016, 060012. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Manju, K.; Muralikrishna, B.; Parnaik, V.K. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J. Cell Sci. 2006, 119, 2704–2714. [Google Scholar] [CrossRef] [Green Version]
- Perepelina, K.; Dmitrieva, R.; Ignatieva, E.; Borodkina, A.; Kostareva, A.; Malashicheva, A. Lamin A/C mutation associated with lipodystrophy influences adipogenic differentiation of stem cells through interaction with Notch signaling. Biochem. Cell Biol. 2018, 96, 342–348. [Google Scholar] [CrossRef]
- Perepelina, K.; Klauzen, P.; Kostareva, A.; Malashicheva, A. Tissue-Specific Influence of Lamin A Mutations on Notch Signaling and Osteogenic Phenotype of Primary Human Mesenchymal Cells. Cells 2019, 8, 266. [Google Scholar] [CrossRef] [Green Version]
- Bechert, K.; Lagos-Quintana, M.; Harborth, J.; Weber, K.; Osborn, M. Effects of expressing lamin A mutant protein causing Emery-Dreifuss muscular dystrophy and familial partial lipodystrophy in HeLa cells. Exp. Cell Res. 2003, 286, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C Expression Is a Marker of Mouse and Human Embryonic Stem Cell Differentiation. Stem Cells 2006, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; O’Rourke, J.R.; Sutherland, L.B.; Bezprozvannaya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Vashisht, A.A.; O’Rourke, J.; Corbel, S.Y.; Moran, R.; Romero, A.; Miraglia, L.; Zhang, J.; Durrant, E.; Schmedt, C.; et al. The microprotein Minion controls cell fusion and muscle formation. Nat. Commun. 2017, 8, 15664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pownall, M.E.; Gustafsson, M.K.; Emerson, C.P. Myogenic Regulatory Factors and the Specification of Muscle Progenitors in Vertebrate Embryos. Annu. Rev. Cell Dev. Biol. 2002, 18, 747–783. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Soufi, A.; Dalton, S. Cycling through developmental decisions: How cell cycle dynamics control pluripotency, differentiation and reprogramming. Development 2016, 143, 4301–4311. [Google Scholar] [CrossRef] [Green Version]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements Downloaded from. J. Biol. Chem. 2017, 7189. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, A.; Mozzetta, C.; Pegoli, G.; Lucini, F.; Valsoni, S.; Rosti, V.; Petrini, C.; Cortesi, A.; Gregoretti, F.; Antonelli, L.; et al. Dysfunctional polycomb transcriptional repression contributes to lamin A/C–dependent muscular dystrophy. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [Green Version]
- Malashicheva, A.; Bogdanova, M.; Zabirnyk, A.; Smolina, N.; Ignatieva, E.; Freilikhman, O.; Fedorov, A.; Dmitrieva, R.; Sjöberg, G.; Sejersen, T.; et al. Various lamin A/C mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol. Genet. Metab. 2015, 115, 118–127. [Google Scholar] [CrossRef]
- Spuler, S.; Kalbhenn, T.; Zabojszcza, J.; Van Landeghem, F.K.H.; Ludtke, A.; Wenzel, K.; Koehnlein, M.; Schuelke, M.; Lüdemann, L.; Schmidt, H.H. Muscle and nerve pathology in Dunnigan familial partial lipodystrophy. Neurology 2007, 68, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Battersby, B.J.; Hansford, R.G.; Moyes, C.D. Interactions between bioenergetics and mitochondrial biogenesis. Biochim. Biophys. Acta Bioenerg. 1998, 1365, 522–530. [Google Scholar] [CrossRef] [Green Version]
- Wagatsuma, A.; Sakuma, K. Mitochondria as a potential regulator of myogenesis. Sci. World J. 2013, 2013, 593267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, F.; Di Girolamo, D.; Mella, S.; Yennek, S.; Chatre, L.; Ricchetti, M.; Tajbakhsh, S. Distinct metabolic states govern skeletal muscle stem cell fates during prenatal and postnatal myogenesis. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Rasmussen, L.J. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 1925, 2012. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Martin, C.; Maya-Mendoza, A.; Tang, C.W.; Lovrić, J.; Sims, P.F.G.; Jackson, D.A. Reduced expression of lamin A/C results in modified cell signaling and metabolism coupled with changes in expression of structural proteins. J. Proteome Res. 2009, 8, 5196–5211. [Google Scholar] [CrossRef]
- Magagnotti, C.; Bachi, A.; Zerbini, G.; Fattore, E.; Fermo, I.; Riba, M.; Previtali, S.C.; Ferrari, M.; Andolfo, A.; Benedetti, S. Protein profiling reveals energy metabolism and cytoskeletal protein alterations in LMNA mutation carriers. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 970–979. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| hLMNA_R482L forward | TTACC(G/T)GTTCCCACCAAAGTTCACCCTGAAGG * |
| hLMNA_R482L reverse | GGAAC(C/A)GGTAAGTCAGCAAGGGATCATCTCCA * |
| hLMNA_G232E forward | CTGGTGGAGATTGACAATG(G/A)GAAGCAGCGTGAGTTTGAG * |
| hLMNA_G232E reverse | CTCAAACTCACGCTGCTTC(C/T)CATTGTCAATCTCCACCAG * |
| Primer Name | Primer Sequence (5′-3′) |
|---|---|
| hLMNA_R482L seq-forward | GCTGGTCGAGTACCAGGAGCTTCTGGACATCA |
| hLMNA_R482L seq-reverse | GCCGTAGGCAGGCTGTTCCCGCAGCCCCAGGT |
| hLMNA_G232E seq-forward | GGATGAGATGCTGCGGCGG |
| hLMNA_G232E seq-reverse | GCTGGGCAGAGAGGCTGTCG |
| Number of Genes in Overlap | |||
|---|---|---|---|
| WT vs. G232E | WT vs. R482L | R482L vs. G232E | |
| Myogenesis (Hallmark database) | 29 | 15 | 14 |
| Muscle structure development | 54 | 16 | 23 |
| Muscle system process | 48 | 20 | 24 |
| Muscle contraction | 41 | 18 | 21 |
| Striated muscle cell differentiation | 38 | 11 | 18 |
| Muscle cell differentiation | 41 | 11 | 19 |
| Muscle cell development | 30 | 10 | 14 |
| Myofibril assembly | 20 | 8 | 11 |
| Cellular component assembly involved in morphogenesis | 20 | 8 | 11 |
| Striated muscle contraction | 20 | 10 | 10 |
| Anatomical structure formation involved in morphogenesis | 39 | 14 | 20 |
| Muscle filament sliding | 13 | 8 | 7 |
| Actomyosin structure organization | 20 | 8 | 11 |
| Actin filament based process | 31 | ns | 15 |
| Sarcomere organization | 13 | ns | 7 |
| Myotube differentiation | 16 | ns | 9 |
| Muscle fiber development | 14 | ns | 9 |
| Actin mediated cell contraction | ns | 8 | ns |
| Regulation of muscle contraction | ns | 7 | ns |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ignatieva, E.V.; Ivanova, O.A.; Komarova, M.Y.; Khromova, N.V.; Polev, D.E.; Kostareva, A.A.; Sergushichev, A.; Dmitrieva, R.I. LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile. Genes 2020, 11, 1057. https://doi.org/10.3390/genes11091057
Ignatieva EV, Ivanova OA, Komarova MY, Khromova NV, Polev DE, Kostareva AA, Sergushichev A, Dmitrieva RI. LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile. Genes. 2020; 11(9):1057. https://doi.org/10.3390/genes11091057
Chicago/Turabian StyleIgnatieva, Elena V., Oksana A. Ivanova, Margarita Y. Komarova, Natalia V. Khromova, Dmitrii E. Polev, Anna A. Kostareva, Alexey Sergushichev, and Renata I. Dmitrieva. 2020. "LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile" Genes 11, no. 9: 1057. https://doi.org/10.3390/genes11091057
APA StyleIgnatieva, E. V., Ivanova, O. A., Komarova, M. Y., Khromova, N. V., Polev, D. E., Kostareva, A. A., Sergushichev, A., & Dmitrieva, R. I. (2020). LMNA Mutations G232E and R482L Cause Dysregulation of Skeletal Muscle Differentiation, Bioenergetics, and Metabolic Gene Expression Profile. Genes, 11(9), 1057. https://doi.org/10.3390/genes11091057