
Prevalence and Clinical Characteristics of Hearing Loss Caused by MYH14 Variants


 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Variant Analysis
2.3. Clinical Evaluation
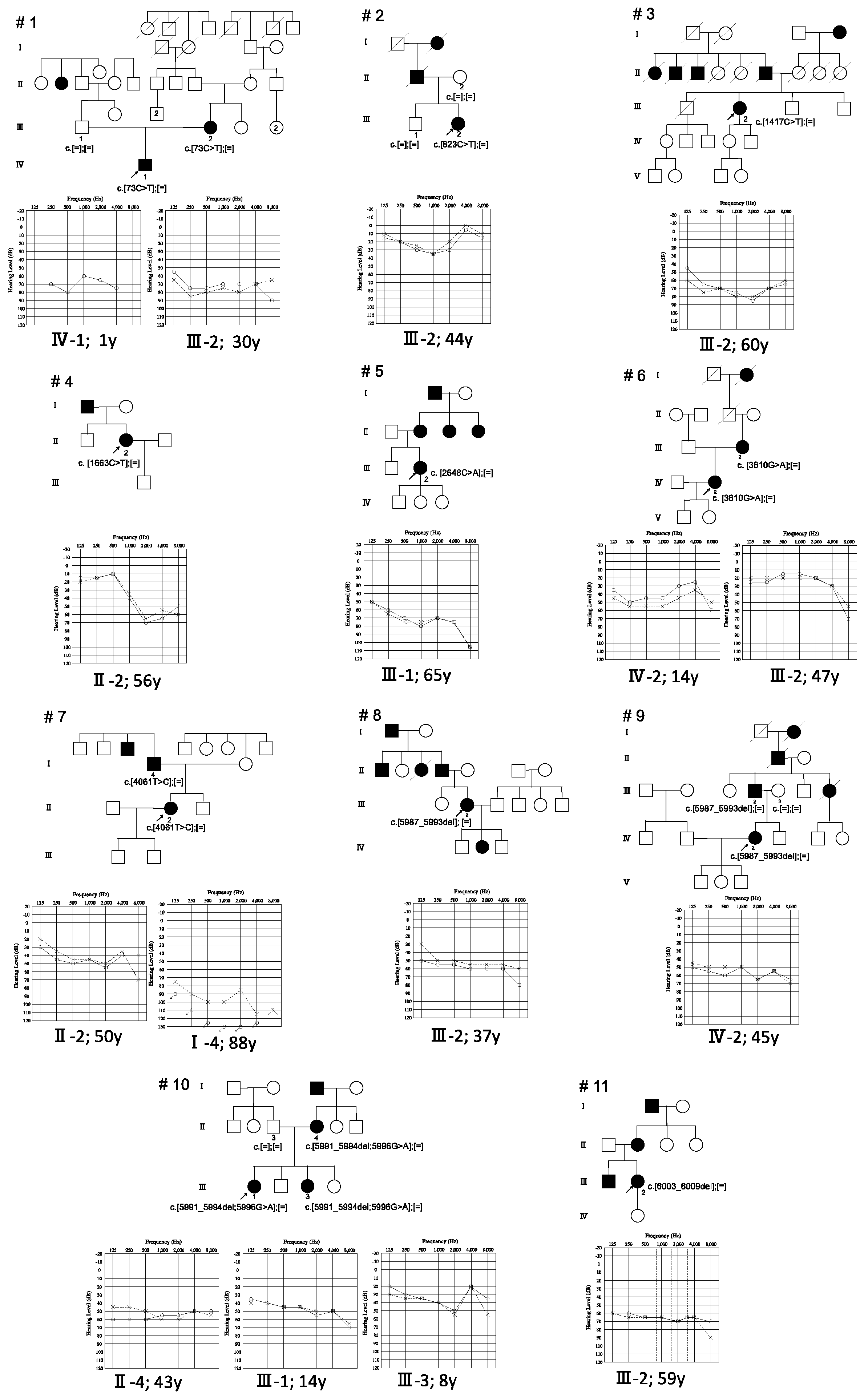
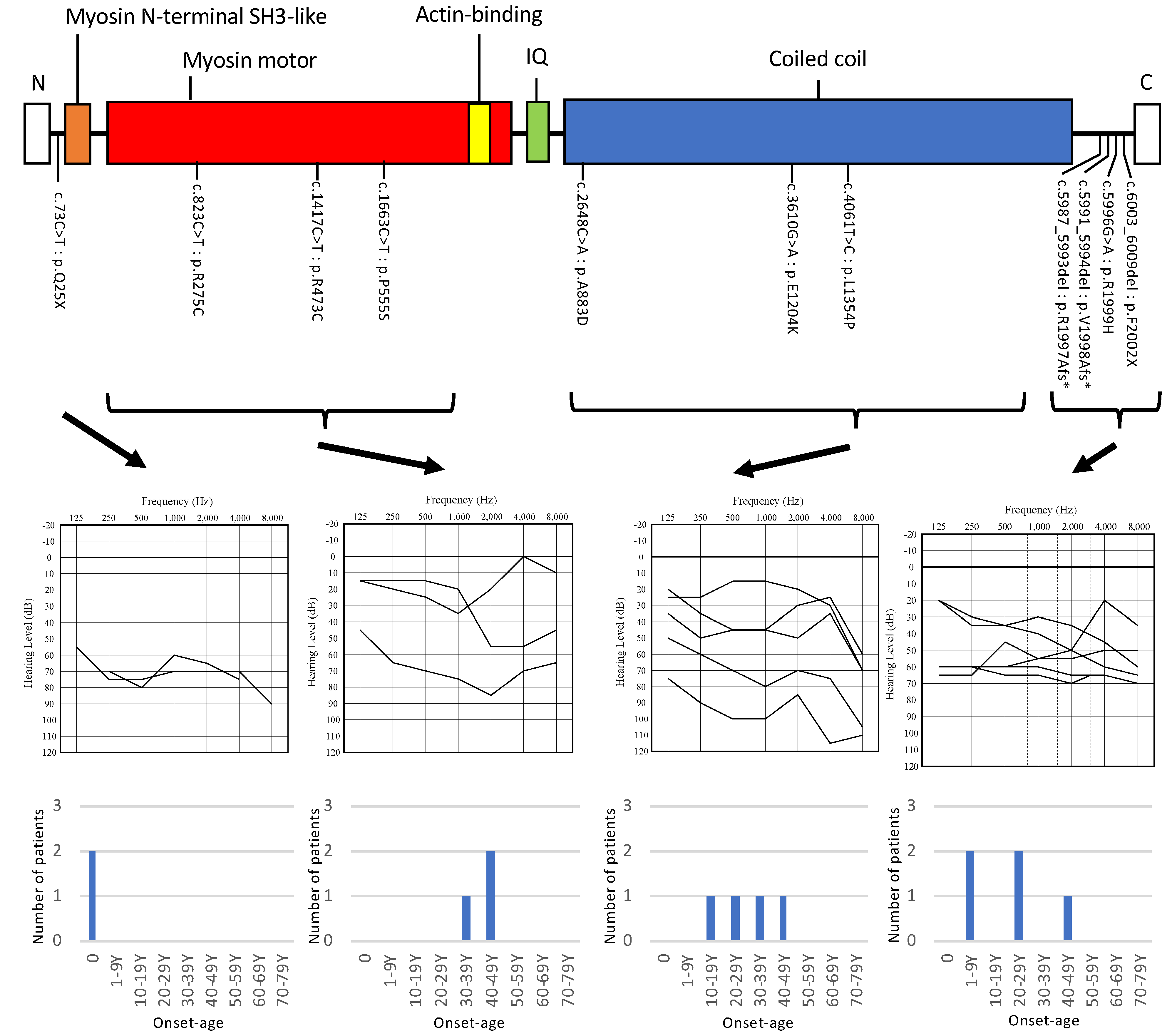
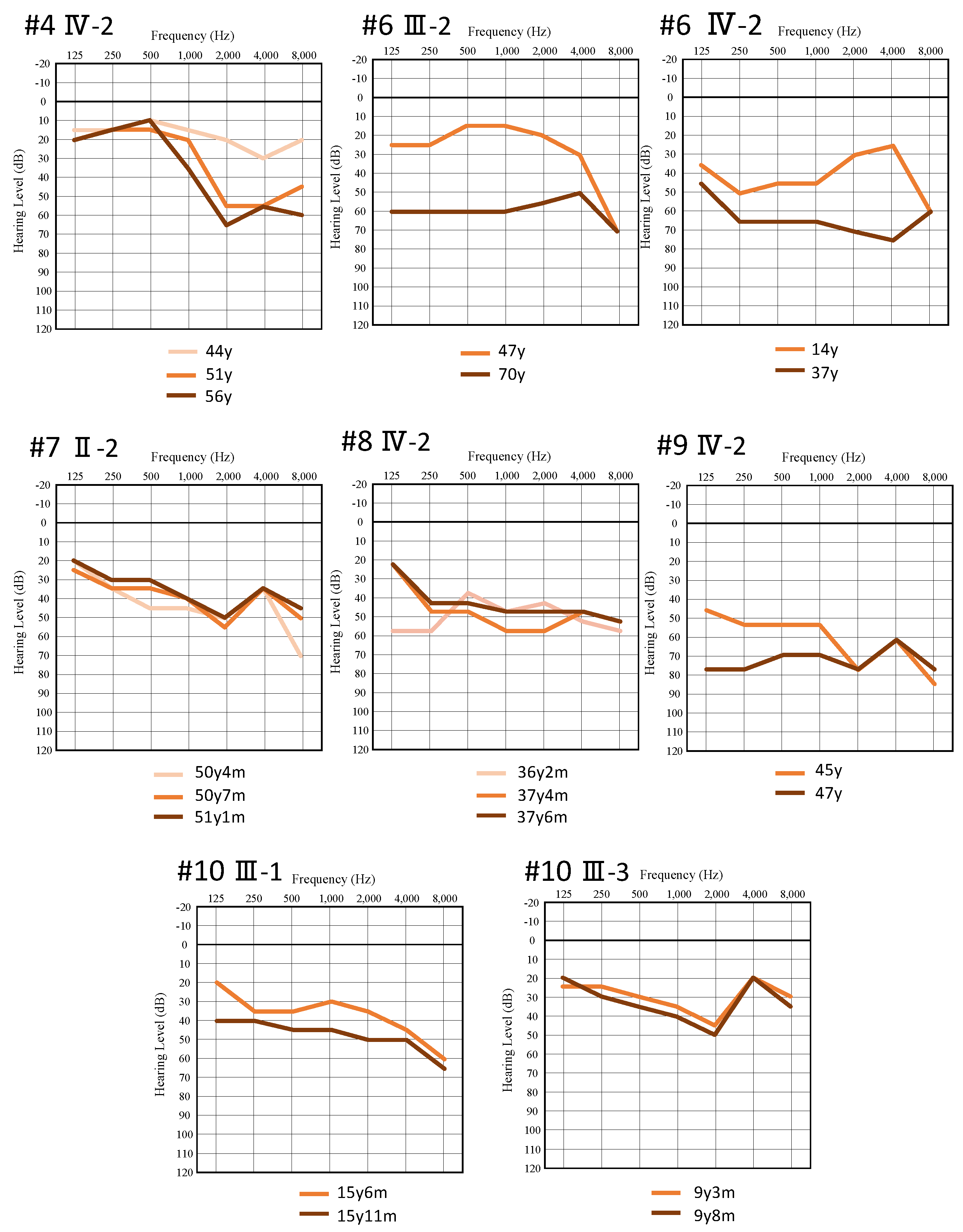
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hereditary Hearing Loss Homepage. Available online: http://hereditaryhearingloss.org/ (accessed on 30 December 2019).
- Smith, R.J.H.; Bale, J.F.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Donaudy, F.; Snoeckx, R.; Pfister, M.; Zenner, H.P.; Blin, N.; Di Stazio, M.; Ferrara, A.; Lanzara, C.; Ficarella, R.; Declau, F.; et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 2004, 74, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.J.; Kim, A.R.; Han, J.H.; Lee, C.; Oh, D.Y.; Choi, B.Y. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. J. Gene Med. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Pfister, M.; Blin, N.; Zenner, H.P.; Pusch, C.M.; Smith, R.J. Genetic heterogeneity of deafness phenotypes linked to DFNA4. Am. J. Med. Genet. A 2013, 139, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, Y.I.; Nishio, S.Y.; Usami, S.I. Comprehensive Genetic Analysis of Japanese Autosomal Dominant Sensorineural Hearing Loss Patients. PLoS ONE 2016, 11, e0166781. [Google Scholar] [CrossRef]
- Shearer, A.E.; Black-Ziegelbein, E.A.; Hildebrand, M.S.; Eppsteiner, R.W.; Ravi, H.; Joshi, S.; Guiffre, A.C.; Sloan, C.M.; Happe, S.; Howard, S.D.; et al. Advancing genetic testing for deafness with genomic technology. J. Med. Genet. 2013, 50, 627–634. [Google Scholar] [CrossRef]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Dong, C.; Wang, Q.; Zhong, Z.; Qi, Y.; Ke, X.; Liu, Y. Targeted Next-Generation Sequencing Successfully Detects Causative Genes in Chinese Patients with Hereditary Hearing Loss. Genet. Test. Mol. Biomark. 2016, 20, 660–665. [Google Scholar] [CrossRef]
- Qing, J.; Yan, D.; Zhou, Y.; Liu, Q.; Wu, W.; Xiao, Z.; Liu, Y.; Liu, J.; Du, L.; Xie, D.; et al. Whole-Exome Sequencing to Decipher the Genetic Heterogeneity of Hearing Loss in a Chinese Family with Deaf by Deaf Mating. PLoS ONE 2014, 9, e109178. [Google Scholar] [CrossRef] [Green Version]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; Ende, J.V.D.; Boudewyns, A.; Leenheer, E.D.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef]
- Shearer, A.E.; Eppsteiner, R.W.; Booth, K.T.; Ephraim, S.S.; Gurrola, J., 2nd; Simpson, A.; Black-Ziegelbein, E.A.; Joshi, S.; Ravi, H.; Giuffre, A.C.; et al. Utilizing Ethnic-Specific Differences in Minor Allele Frequency to Recategorize Reported Pathogenic Deafness Variants. Am. J. Hum. Genet. 2014, 95, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelhoda, M.; Faquih, T.; El-kalioby, M.; Alkuraya, F.S. Revisiting the morbid genome of Mendelian disorders. Genome Biol. 2016, 17, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.K.D.; Kim, A.R.; Park, K.T.; Kim, S.Y.; Kim, M.Y.; Nam, J.-Y.; Woo, S.J.; Oh, S.-H.; Park, W.-Y. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet. Med. 2015, 17, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.-H.; Chang, P.-Y.; Chang, S.-C.; Lu, J.-J.; Wu, C.-M. Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS ONE 2019, 14, e0211261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, M.; Nishio, S.; Ikeda, T.; Fukushima, K.; Usami, S. Massively Parallel DNA Sequencing Successfully Identifies New Causative Mutations in Deafness Genes in Patients with Cochlear Implantation and EAS. PLoS ONE 2013, 8, e75793. [Google Scholar]
- Choi, B.O.; Hee Kang, S.; Hyun, Y.S.; Kanwal, S.; Park, S.W.; Koo, H.; Kim, S.B.; Choi, Y.C.; Yoo, J.H.; Kim, J.W.; et al. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Hum. Mutat. 2011, 32, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Lee, S.; Park, H.J.; Kang, T.H.; Sagong, B.; Baek, J.I.; Oh, S.K.; Choi, J.Y.; Lee, K.Y.; Kim, U.K. Genetic association of MYH genes with hereditary hearing loss in Korea. Gene 2016, 591, 177–182. [Google Scholar] [CrossRef]
- Vona, B.; Müller, T.; Nanda, I.; Neuner, C.; Hofrichter, A.H.H.; Schröder, J.; Bartsch, O.; Läßig, A.; Keilmann, A.; Schraven, S.; et al. Open Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations. Genet. Med. 2014, 16, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Iyadurai, S.; Arnold, W.D.; Kissel, J.T.; Ruhno, C.; McGovern, V.L.; Snyder, P.J.; Prior, T.W.; Roggenbuck, J.; Burghes, A.H.; Kolb, S.J. Variable phenotypic expression and onset in MYH14 distal HMN phenotype in a large, multigenerational North American family. Muscle Nerve 2017, 176, 139–148. [Google Scholar]
- Almutawa, W.; Smith, C.; Sabouny, R.; Smit, R.B.; Zhao, T.; Wong, R.; Lee-Glover, L.; Desrochers-Goyette, J.; Ilamathi, H.S.; Suchowersky, O.; et al. The R941L mutation in MYH14 disrupts mitochondrial fission and associates with peripheral neuropathy. EBiolMedicine 2019, 45, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Seco, C.Z.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; Wijs, I.W.; Admiraal, R.J.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in the Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef] [Green Version]
- Moteki, H.; Azaiez, H.; Booth, K.T.; Shearer, A.E.; Sloan, C.M.; Kolbe, D.L.; Nishio, S.Y.; Hattori, M.; Usami, S.I.; Smith, R.J. Comprehensive genetic testing with ethnic-specific filtering by allele frequency in a Japanese hearing-loss population. Clin. Genet. 2017, 89, 466–472. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, Y.; Zhang, F.; Fan, Z.; Bai, X.; Wang, H. A novel MYH14 mutation in a Chinese family with autosomal dominant nonsyndromic hearing loss. BMC Med. Genet. 2020, 21, 154. [Google Scholar] [CrossRef]
- Maekawa, K.; Nishio, S.; Abe, S.; Goto, S.I.; Honkura, Y.; Iwasaki, S.; Kanda, Y.; Kobayashi, Y.; Oka, S.; Okami, M.; et al. Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes 2019, 10, 735. [Google Scholar] [CrossRef] [Green Version]
- The Exome Aggregation Consortium Database (ExAC). Available online: http://exac.broadinstitute.org/ (accessed on 1 July 2019).
- ToMMo 3.5KJPN-Integrative Japanese Genome Variation Database. Available online: https://jmorp.megabank.tohoku.ac.jp/201905/ (accessed on 1 July 2019).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Mazzoli, M.; Camp, G.V.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the Description of Genetic and Audiological Data for Families with Nonsyndromic Hereditary Hearing Impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar]
- Naito, T.; Nishio, S.; Iwasa, Y.; Yano, T.; Kumakawa, K.; Abe, S.; Ishikawa, K.; Kojima, H.; Namba, A.; Oshikawa, C.; et al. Comprehensive Genetic Screening of KCNQ4 in a Large Autosomal Dominant Nonsyndromic Hearing Loss Cohort: Genotype-Phenotype Correlations and a Founder Mutation. PLoS ONE 2013, 8, e63231. [Google Scholar] [CrossRef]
- Kitano, T.; Miyagawa, M.; Nishio, S.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-34based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017, 12, e0177636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasukawa, R.; Moteki, H.; Nishio, S.; Ishikawa, K.; Abe, S.; Honkura, Y.; Misako Hyogo, M.; Mihashi, R.; Ikezono, T.; Shintani, T.; et al. The prevalence and clinical characteristics of TECTA-associated autosomal dominant hearing loss. Genes 2019, 10, 744. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Miyagawa, M.; Nishio, S.; Moteki, H. WFS1 mutation screening in a large series of Japanese hearing loss patients: Massively parallel DNA sequencing-based analysis. PLoS ONE 2018, 13, e0193359. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Day, T.F.; Nishio, S.; Moteki, H.; Miyagawa, M.; Morita, S.; Izumi, S.; Ikezono, T.; Abe, S.; Nakayama, J.; et al. Clinical characteristics and in vitro analysis of MYO6 variants causing late-onset progressive hearing loss. Genes 2020, 11, 273. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, H.; Moteki, H.; Day, T.; Nishio, S.; Murata, T.; Ikezono, T.; Takeda, H.; Abe, S.; Iwasaki, S.; Takahashi, M.; et al. Novel ACTG1 mutations in patients identified by massively parallel DNA sequencing cause progressive hearing loss. Sci. Rep. 2020, 10, 7056. [Google Scholar] [CrossRef]
- Shinagawa, J.; Moteki, H.; Nishio, S.; Ohyama, K.; Otsuki, K.; Iwasaki, S.; Masuda, S.; Oshikawa, C.; Ohta, Y.; Arai, Y.; et al. Prevalence and clinical features of hearing loss caused by EYA4 variants. Sci. Rep. 2020, 10, 3662. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Amino Acid Change | Gender | Onset | Progression of HL | Age | Audiometric Configuration | PTA (Better-Hearing Ear, dB) | Severity | Vestibular Symptoms | Intervention | Newborn Hearing Screening | Family No. | Patient No. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.73C > T | p.Q25X | F | 0 s | No | 30 Y | Flat | 71.25 | Severe | No | HA | refer | 1 | Ⅲ-2 |
| c.73C > T | p.Q25X | M | 0 | n/a | 1 Y | Flat | 70 | Moderate | n/a | n/a | n/a | 1 | Ⅳ-1 |
| c.823C > T | p.R275C | F | 41 Y | Yes | 44 Y | U-shaped | 20 | Mild | No | n/a | n/a | 2 | Ⅲ-2 |
| c.1417C > T | p.R473C | F | 35 Y | Yes | 60 Y | Flat | 75 | Severe | n/a | n/a | n/a | 3 | Ⅲ-2 |
| c.1663C > T | p.P555S | F | 44 Y | Yes | 56 Y | Steeply sloping | 41.25 | Moderate | No | HA | n/a | 4 | Ⅱ-3 |
| c.2648C > A | p.A883D | F | 44 Y | n/a | 65 Y | Gently sloping | 73.75 | Moderate | Yes | n/a | n/a | 5 | Ⅲ-1 |
| c.3610G > A | p.E1204K | F | 12 Y | Yes | 14 Y | Low-frequency ascending | 47.5 | Moderate | No | HA | n/a | 6 | III-1 |
| c.3610G > A | p.E1204K | F | n/a | Yes | 47 Y | Flat | 20 | Mild | No | n/a | n/a | 6 | Ⅱ-3 |
| c.4061T > C | p.L1354P | M | 20 s | Yes | 83 Y | Flat | 100 | Profound | n/a | n/a | n/a | 7 | I-4 |
| c.4061T > C | p.L1354P | F | 30 s | Yes | 50 Y | Flat | 43.75 | Moderate | No | HA | n/a | 7 | II-2 |
| c.5987_5993del | p.R1997Afs * | F | 20 Y | Yes | 37 Y | Flat | 53.75 | Moderate | Yes | HA | n/a | 8 | Ⅲ-2 |
| c.5987_5993del | p.R1997Afs * | F | 20 s | Yes | 45 Y | Flat | 55 | Moderate | No | HA | n/a | 9 | Ⅳ-2 |
| c.5987_5993del | p.R1997Afs * | M | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | 9 | Ⅲ-2 |
| c.[5991_5994del; 5996G > A] | p.[V1998Afs *; R1999H] | F | n/a | n/a | 43 Y | Flat | 55 | Moderate | n/a | n/a | n/a | 10 | Ⅱ-4 |
| c.[5991_5994del; 5996G > A] | p.[V1998Afs *; R1999H] | M | 6 Y | Yes | 14 Y | Flat | 47.5 | Moderate | No | HA | pass | 10 | Ⅲ-1 |
| c.[5991_5994del; 5996G > A] | p.[V1998Afs *; R1999H] | F | 3 Y | Yes | 8 Y | U-shaped | 29 | Mild | No | HA | pass | 10 | Ⅲ-3 |
| c.6003_6009del | p.F2002X | F | 41 Y | Yes | 59 Y | Flat | 66.25 | Moderate | No | n/a | n/a | 11 | Ⅲ-2 |
| Nucleotide Change | Exon | Amino Acid Change | Domain | SIFT | PP2 | Mut Taster | Mut Assessor | Revel | CADD Phred | Allele Frequency in In-house Controls | MAF in ExAC03 | MAF in ToMMo (4.7kJPN) | ACMG Criteria | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.73C > T | 2 | p.Q25X | Myosin, N-terminal | A | 36 | 0 | 0 | 0 | Likely Pathogenic | Donaudy et al., 2004 [3] | ||||
| c.823C > T | 8 | p.R275C | Myosin head, motor domain | D | D | D | D | 0.648 | 34 | 0 | 0.000008252 | 0 | Uncertain Significance | Iwasa et al., 2016 [6] |
| c.1417C > T | 13 | p.R473C | Myosin head, motor domain | D | P | D | M | 0.648 | 34 | 0 | 2.81 × 10−5 | 0 | Uncertain Significance | This study |
| c.1663C > T | 15 | p.P555S | Myosin head, motor domain | D | D | D | M | 0.701 | 26.1 | 0 | 0 | 0 | Uncertain Significance | This study |
| c.2648C > A | 22 | p.A883D | Myosin tail | D | D | D | M | 0.692 | 28 | 0 | 0 | 0 | Uncertain Significance | This study |
| c.3610G > A | 28 | p.E1204K | Myosin tail | T | B | D | L | 0.678 | 24.7 | 0 | 2.29 × 10−5 | 0 | Uncertain Significance | This study |
| c.4061T > C | 31 | p.L1354P | Myosin tail | D | D | D | M | 0.813 | 28.4 | 0 | 0 | 0 | Uncertain Significance | This study |
| c.5987_5993del | p.R1997Afs | 0 | 0 | 0 | Uncertain Significance | This study | ||||||||
| c.[5991_5994del; 5996G > A] | 43 | p.[V1998Afs; R1999H] | 0 | 0 | 0 | Uncertain Significance | This study | |||||||
| D | P | N | L | 0.251 | 24.1 | 0 | 3.35 × 10−5 | 0 | Uncertain Significance | This study | ||||
| c.6003_6009del | 43 | p.F2002X | 0 | 0 | 0 | Uncertain Significance | This study |
| Nucleotide Change | Amino Acid Change | Gender | Onset | Progression of HL | Age | Audiometric Configuration | PTA (Better-Hearing Ear) | Severity | Vestibular Symptoms | Intervention | Newborn Hearing Screening | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.20C > A | p.S7X | n/a | 10 or 20 | Yes | n/a | n/a | n/a | Severe to profound in 40 years | n/a | n/a | n/a | Donaudy et al., 2004 [3] |
| c.73C > T | p.Q25X | F | 0 Y | Yes | 7 M | n/a | n/a | n/a | n/a | n/a | n/a | Kim et al., 2017 [4] |
| F | n/a | n/a | 33 Y | Flat | 82.5 | Severe | n/a | n/a | n/a | |||
| c.359C > T | p.S120L | n/a | n/a | n/a | 28 Y | Flat | 53.8 | Moderate | n/a | n/a | n/a | Yang et al., 2005 [5] |
| n/a | n/a | n/a | 33 Y | Flat | 65 | Moderate | n/a | n/a | n/a | |||
| n/a | n/a | n/a | 35 Y | Flat | 76.3 | Moderate | n/a | n/a | n/a | |||
| n/a | n/a | n/a | 63 Y | Flat | 72.5 | Moderate | n/a | n/a | n/a | |||
| c.505G > A | p.E169K | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sloan-Heggen et al., 2016 [8] |
| c.526G > A | p.A176T | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Chen et al., 2016 [9] |
| c.541G > A | p.A181T | F | The first decade | n/a | n/a | U-sharped | 73.75 | Moderate | n/a | n/a | n/a | Qing et al., 2014 [10] |
| M | The first decade | n/a | n/a | Flat | 71.3 | Moderate | n/a | n/a | n/a | |||
| F | The first decade | n/a | n/a | U-sharped | n/a | Severe | n/a | n/a | n/a | |||
| c.572A > G | p.D191G | M | Congenital or prelingual | No? | 5 Y | Flat | 71.3 | Moderate | n/a | n/a | n/a | Kim et al., 2017 [4] |
| c.823C > T | p.R275C | F | 41 Y | Yes | 44 Y | U-shaped | 20 | Mild | No | n/a | n/a | Iwasa et al., 2016 [6] |
| c.1049G > A | p.R350Q | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Iwasa et al., 2016 [6] |
| c.1067C > T | p.T356M | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sommen et al., 2016 [11] |
| c.1150G > T | p.G384C | n/a | n/a | n/a | 9 Y | n/a | n/a | Moderate | No | n/a | n/a | Donaudy., 2004 [3] |
| n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Shearer et al.,2014 [12] | ||
| n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Abouelhoda et al., 2016 [13] | ||
| c.1360G > A | p.A454T | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Chen et al., 2016 [9] |
| c.1427G > A | p.R476H | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sloan-Heggen et al., 2016 [8] |
| c.1609G > A | p.D537N | F | n/a | Yes | 8Y | Flat | 40 | Moderate | n/a | n/a | n/a | Kim et al., 2015 [14] |
| c.1625T > G | p.L542R | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sloan-Heggen et al., 2016 [8] |
| c.1919G > A | p.R640Q | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Shearer et al., 2013 [7] |
| c.2089G > A | p.G697S | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Iwasa et al., 2016 [6] |
| c.2203C> G | p.R735C | M | n/a | n/a | n/a | Flat | 115 | Severe | n/a | CI | n/a | Liu et al., 2019 [15] |
| M | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | |||
| c.2299C > A | p.R767S | n/a | n/a | Yes | n/a | n/a | n/a | Mild to moderate | No | n/a | n/a | Donaudy et al., 2004 [3] |
| c.2621T > C | p.L874P | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Chen et al., 2016 [9] |
| c.2692A > C | p.K898Q | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Miyagawa et al., 2013 [16] |
| c.2717C > T | p.T906M | F | The first decade | n/a | n/a | U-shaped | 83 | Severe | n/a | n/a | n/a | Qing et al., 2014 [10] |
| F | The first decade | n/a | n/a | U-shaped | 80 | Severe | n/a | n/a | n/a | |||
| c.2921G > A | p.R974H | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sloan-Heggen et al., 2016 [8] | |
| c.2921G > T | p.R974L | M | n/a | n/a | 52 Y | n/a | n/a | n/a | n/a | n/a | n/a | Choi et al., 2011 [17] |
| M | n/a | n/a | 48 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| F | n/a | n/a | 45 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| F | n/a | n/a | 41 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| M | n/a | n/a | 15 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| c.2921G > T | p.R974L | n/a | 20 Y | Yes | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Iyadurai et al., 2017 [20] |
| c.2921G > A | p.R974L | F | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Almutawa et al., 2019 [21] |
| F | n/a | n/a | 58 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| M | n/a | n/a | 23 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| F | n/a | n/a | 24 Y | n/a | n/a | n/a | n/a | n/a | n/a | |||
| c.3049C > T | p.L1017F | M | n/a | n/a | n/a | n/a | n/a | Mild to moderate | No | n/a | n/a | Donaudy et al., 2004 [3] |
| c.3877G > C | p.E1293Q | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Sommen et al., 2016 [11] |
| c.4903G > A | p.E1635K | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Miyagawa et al., 2013 [16] |
| c.5008C > T | p.R1670C | F | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Vona et al., 2014 [19] |
| c.5020G > A | p.V1674M | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Shearer et al., 2013 [7] |
| c.5176C > T | p.R1726W | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Seco et al., 2017 [22] |
| c.5384G > A | p.R1795H | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | n/a | Moteki et al., 2017 [23] |
| c.5516C > A | p.A1839D | M | 30 s | n/a | 51 Y | Gently sloping | 56.25 | Moderate | No † | n/a | n/a | Wang et al., 2020 [24] |
| M | 30 s | Yes | 45 Y | Gently sloping | 50 | Moderate | n/a † | n/a | n/a | |||
| M | 10 s | Yes | 29 Y | Flat | 56.25 | Moderate | No ‡ | n/a | n/a | |||
| c.5602G > A | p.A1868T | M | n/a | n/a | n/a | n/a | 66.3 | n/a | n/a | n/a | n/a | Kim et al., 2016 [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiramatsu, K.; Nishio, S.-y.; Kitajiri, S.-i.; Kitano, T.; Moteki, H.; Usami, S.-i.; on behalf of the Deafness Gene Study Consortium. Prevalence and Clinical Characteristics of Hearing Loss Caused by MYH14 Variants. Genes 2021, 12, 1623. https://doi.org/10.3390/genes12101623
Hiramatsu K, Nishio S-y, Kitajiri S-i, Kitano T, Moteki H, Usami S-i, on behalf of the Deafness Gene Study Consortium. Prevalence and Clinical Characteristics of Hearing Loss Caused by MYH14 Variants. Genes. 2021; 12(10):1623. https://doi.org/10.3390/genes12101623
Chicago/Turabian StyleHiramatsu, Ken, Shin-ya Nishio, Shin-ichiro Kitajiri, Tomohiro Kitano, Hideaki Moteki, Shin-ichi Usami, and on behalf of the Deafness Gene Study Consortium. 2021. "Prevalence and Clinical Characteristics of Hearing Loss Caused by MYH14 Variants" Genes 12, no. 10: 1623. https://doi.org/10.3390/genes12101623
APA StyleHiramatsu, K., Nishio, S.-y., Kitajiri, S.-i., Kitano, T., Moteki, H., Usami, S.-i., & on behalf of the Deafness Gene Study Consortium. (2021). Prevalence and Clinical Characteristics of Hearing Loss Caused by MYH14 Variants. Genes, 12(10), 1623. https://doi.org/10.3390/genes12101623