1. Introduction
In aerobic organisms, such as animals, fungi, and bacteria, reactive oxygen species (ROS) are produced by normal metabolism and cause oxidative damage to DNA [
1]. Base excision repair (BER) is a highly conserved mechanism from bacteria to humans, and it is essential for repairing endogenous DNA damage, including that caused by alkylation, oxidation, deamination, depurination, and single-strand breaks [
2]. Hypoxia suppresses DNA repair pathways that contribute to genomic instability, and this results in intracellular accumulation of DNA damage [
3,
4,
5,
6]. In addition, ultraviolet (UV)-induced ROS act as powerful oxidants that may cause oxidative DNA damage [
7]. The UV-induced DNA lesions, if not repaired, may cause severe structural distortions in the DNA molecule, thereby affecting important cellular processes, such as DNA replication and transcription, compromising cellular viability and functional integrity and ultimately leading to mutagenesis, tumorigenesis, and cell death [
8,
9].
In the mammalian cell, four different uracil-DNA glycosylase (
UDG) genes have been identified, including
SMUG1,
UNG,
TDG, and
MBD4. The UDG family is a family of enzymes that are crucial to DNA repair; their absence allows some mutations to cause cancer [
10]. Current evidence suggests that, in human cells, TDG and SMUG1 are the major enzymes responsible for the repair of the U:G mismatch caused by spontaneous cytosine deamination, whereas uracil arising in DNA through dUTP misincorporation is mainly dealt with by UNG [
11]. The
SMUG1 (single-strand selective monofunctional uracil-DNA glycosylase) gene plays roles in various molecular functions, such as DNA binding, DNA N-glycosylase activity, and single-strand selective and uracil-DNA N-glycosylase activity. Additionally, SMUG1 is also a key enzyme for repairing 5-hydroxymethyluracil, 5-formyluracil, 5,6-dihydrouracil, alloxan, and other lesions generated during oxidative base damage induced by ionizing radiation and oxygen free radicals [
12]. Although SMUG1 is involved in DNA repair in damaged cells, the functional role of SMUG1 in recognizing the damaged DNA regions in the genome and repairing mechanisms remains unclear.
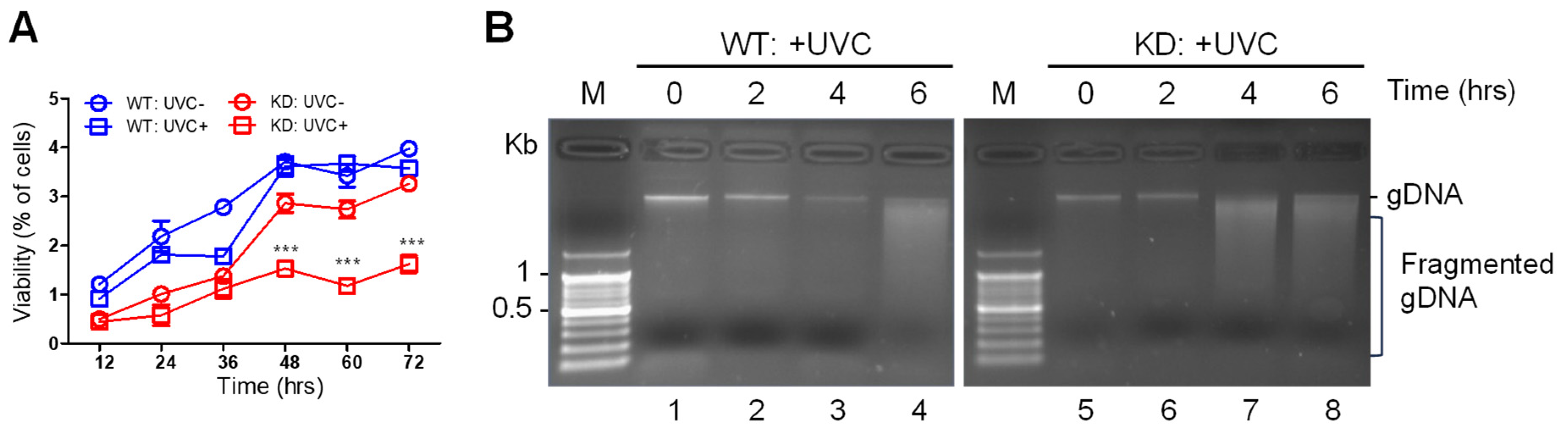
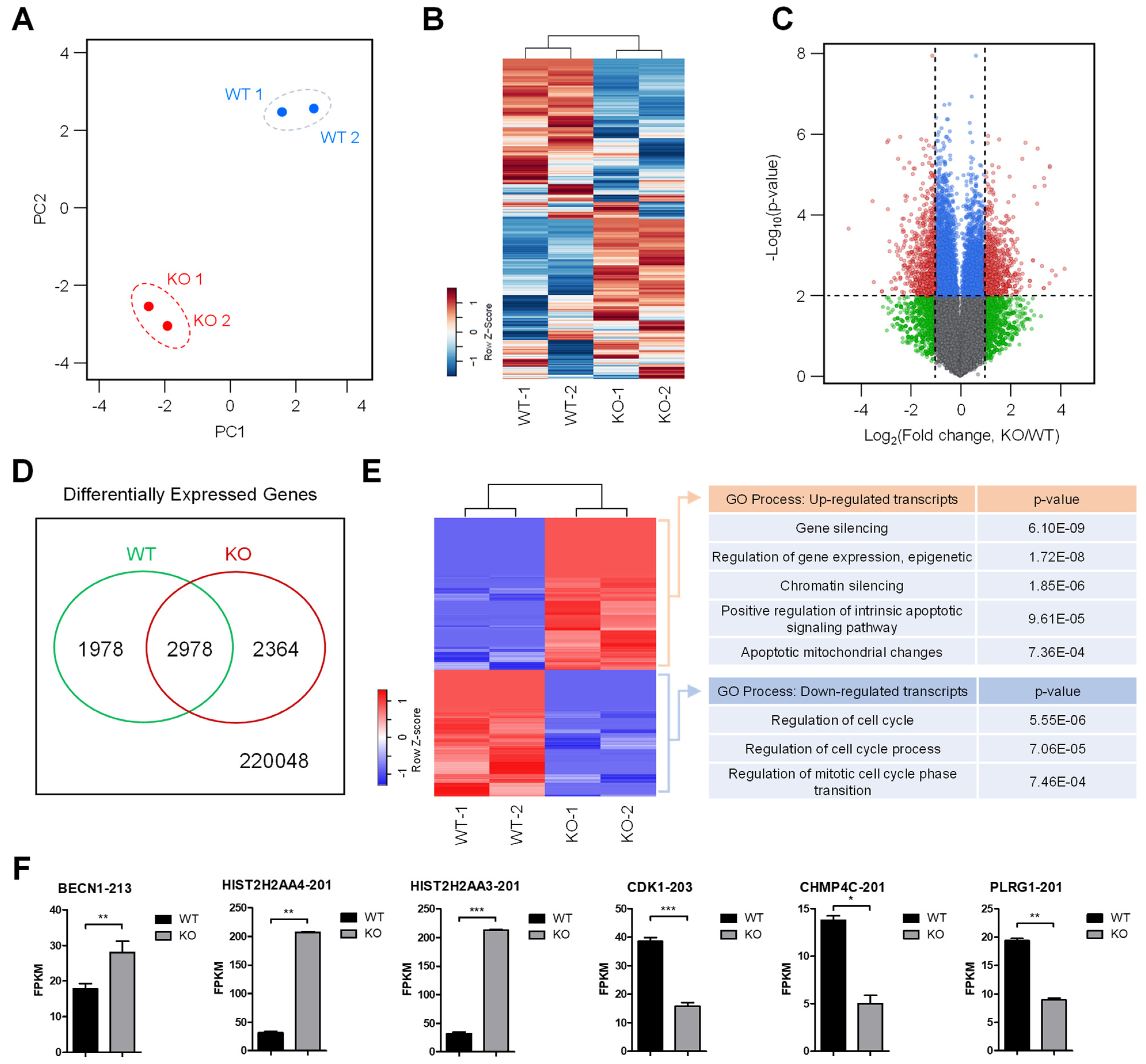
To understand the role of SMUG1 in the regulation of DNA damage responses, we generated knock-down (KD) and knock-out (KO) SMUG1 cell lines using the CRISPR-Cas9 gene-editing system. SMUG1 KO reduces cell proliferation and induces apoptosis. In addition, SMUG1 KD and KO cells were hyposensitive to DNA damage caused by ultraviolet C (UVC) irradiation, and SMUG1 ablation led to apoptosis by delaying the cell cycle. Transcriptome analysis newly revealed that SMUG1 is involved in cell cycle transition and chromatin organization. These results highlight the involvement of SMUG1 in the regulation of DNA damage responses.
2. Materials and Methods
2.1. Cell Cultures and Transfection
All the cell culture reagents used in this study were purchased from Welgene (Seoul, Korea). HepG2 and HEK293T cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in an incubator with 5% CO2 in a humidified atmosphere. For the experiments, the coverslips were treated with 0.1 mg/mL of poly-D lysine for 6 h at room temperature, and they were placed in 100 mm cell culture dishes. Cells were seeded at a density of 2.5 × 106 cells per well in 100 mm cell culture dishes. The other cells in 100 mm cell culture dishes were gently washed with Dulbecco’s phosphate-buffered saline without calcium and magnesium, and then cells were trypsinized. Transient transfection was performed by lipofectamine (Invitrogen, Carlsbad, CA, USA) with different plasmid DNA according to the manufacturer’s instructions.
2.2. Cell Cycle Analysis by the Flowcytometry
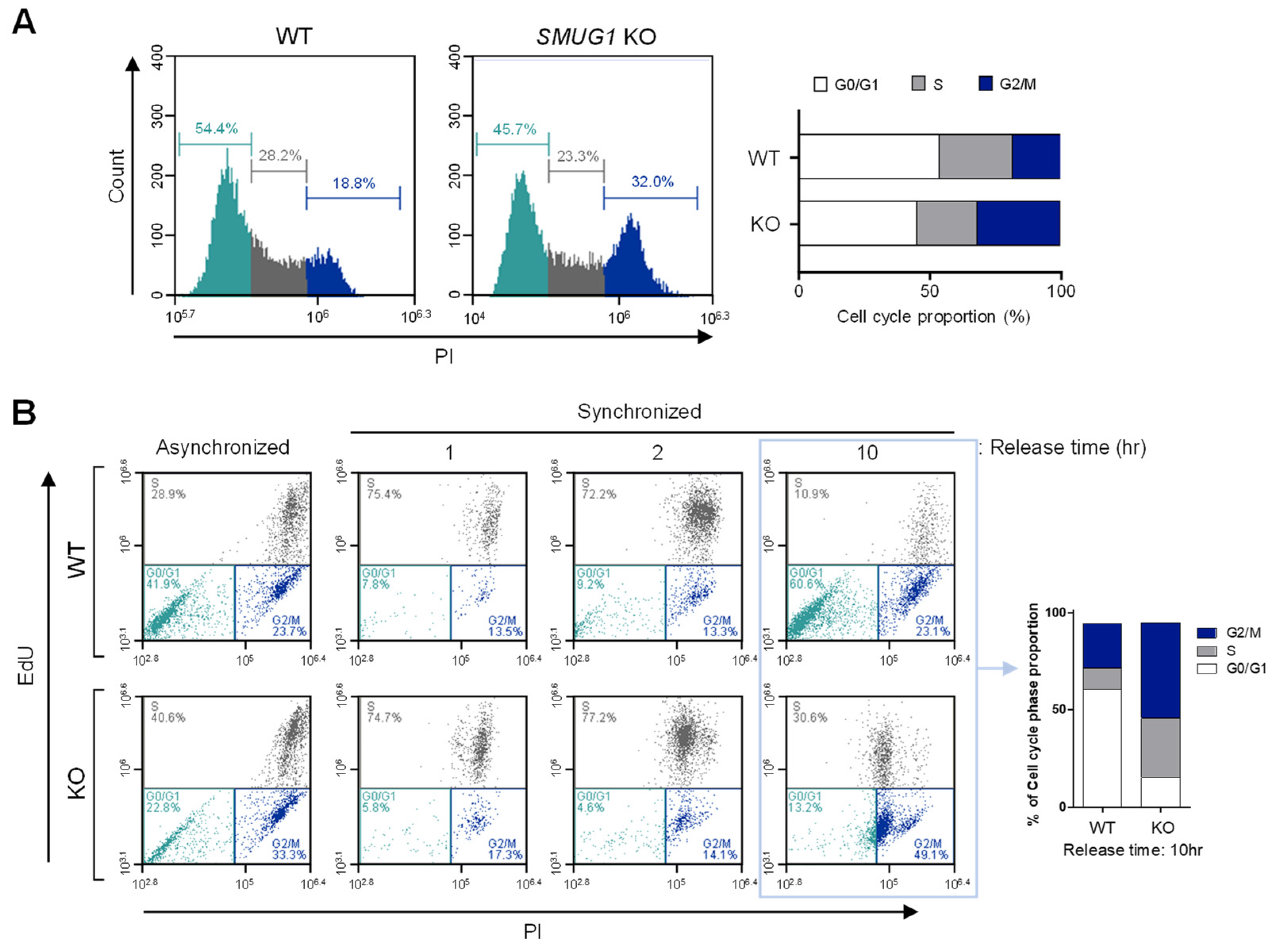
Cell cycle analysis was performed using propidium iodide (PI) staining (Sigma, Burlington, MA, USA). Cells were dissociated with trypsin-EDTA (Welgene, Seoul, Korea) and resuspended with 300 mL of PBS and fixed with 70% ethanol at 4 °C for 1 h. Cell pellets were then resuspended in 0.2 mL of PBS containing 0.25 μg/μL RNase A for 1 h at 37 °C. After that, cells were stained with 10 mL of propidium iodide (PI) solution (1 mg/mL) at room temperature in a dark condition. Finally, 1 × PBS was added to the PI-stained cells and was analyzed by BD Accuri™ C6 Plus (BD FACS, San Jose, CA, USA). At least 10,000 cells were used for each analysis, and the results were displayed as histograms. To investigate S phase progression, we used a double-thymidine block to synchronize cells at the G1 stage and release for the indicated time (0, 1, 2, and 10 h). After releasing, the cells were incubated with EdU for 2 h and stained with both PI and iFluor 488 azide dye. The percentage of cell distribution in the Sub-G1, G0/G1, S, and G2/M phases was measured, and the results were analyzed by the BD Accuri™ C6 Plus software for cell cycle profile.
2.3. Apoptosis Analysis
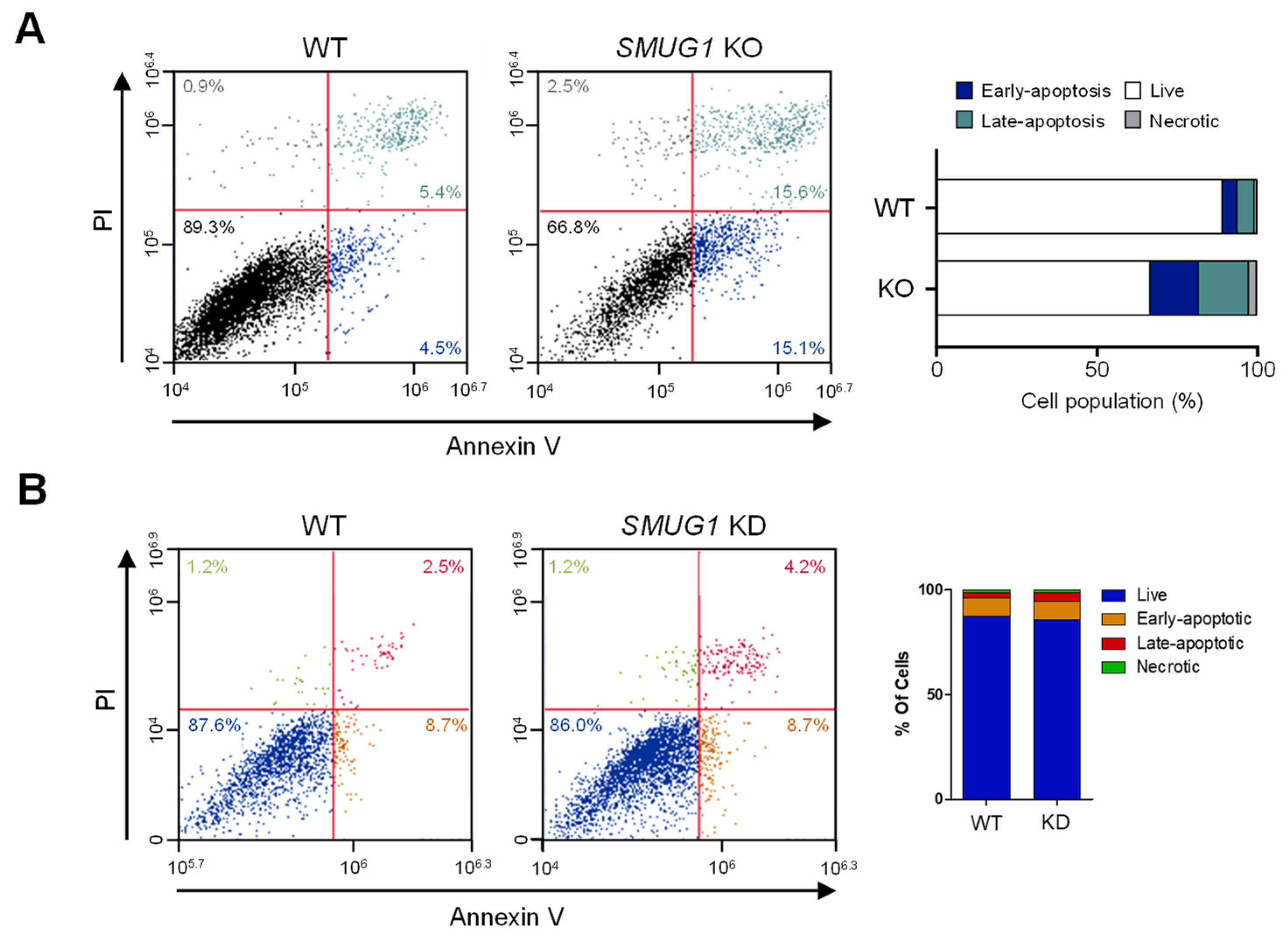
Cell apoptosis analysis was performed by using FITC Annexin-V Apoptosis Detection Kit I (556547; BD Sceince, Franklin Lakes, NJ, USA). Cells were trypsinized and centrifuged at 3000 rpm for 3 min, and the pellets were washed with ice-cold PBS. Then, cells were resuspended with 100 µL of 1 × binding buffer and stained with 5 mL of PI, FITC-Annexin V for 15 min in the dark condition. After PI and FITC-Annexin V staining, cells were mixed with 400 μL of 1 × binding buffer and analyzed by flow cytometry.
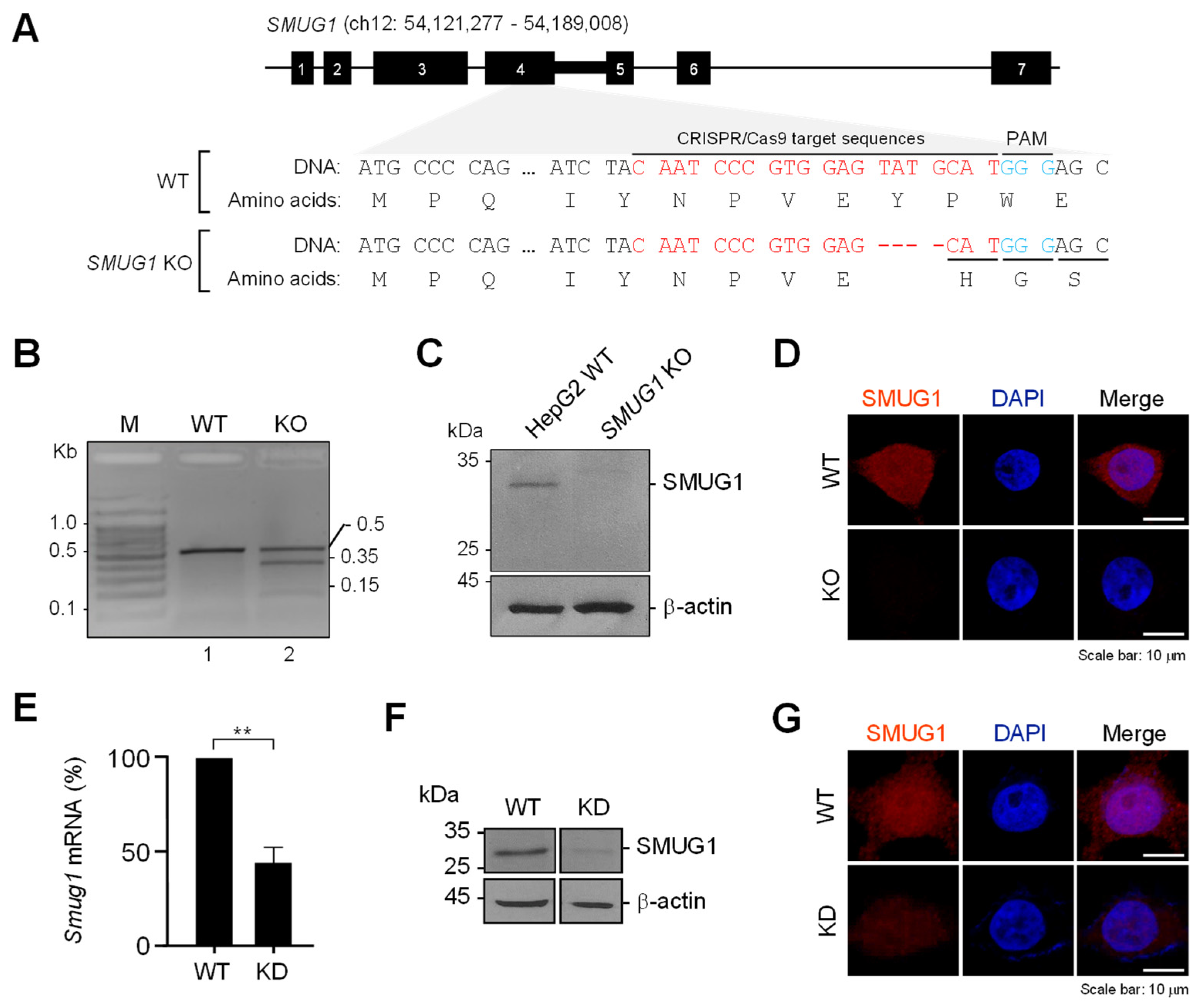
2.4. Generation of SMUG1 Knock-Out (KO) and Knock-Down (KD) Stable Cell Lines
The SMUG1 sgRNA target sequence was designed using the sgRNA prediction program (
http://crispor.tefor.net). Each oligo was phosphorylated and annealed using T4 polynucleotide kinase (NEB, Ipswich, MA, USA). The lenti-sgRNA puro vector (Addgene, #104990) was digested with BsmBI (NEB, Ipswich, MA, USA) at 55 °C for 1 h. The digested vector was separated by gel electrophoresis and extracted using a Gel/PCR purification kit (Favorgen, Ping-Tung, Taiwan). The digested vector was checked with Nanodrop to confirm good DNA quality. The annealed oligo was ligated into the BsmBI-digested lenti-sgRNA puro vector at 4 °C overnight. For the transformation of bacteria, ligation reaction was mixed with 100 mL competent DH5a (RBC, #RH617). Sequencing of cloned lenti-sgRNA puro was performed using U6 promoter sequence (5′-GGG CAG GAA GAG GGC CTA T-3′) by Cosmo genetech. The construct was transfected to HepG2 cells and treated with 1 mg/ mL of puromycin for 4 to 7 days. Selected cells were recovered with Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin for approximately 5 days.
To create the shRNA expression vector targeting for human SMUG1 gene, targeted DNA oligonucleotides were sub-cloned into pLKO.1-puro lentiviral vector (Addgene, Watertown, MA, USA). To produce virus particles, HEK293T cells were co-transfected with the plasmids encoding VSV-G lentiviral vector, packaging plasmid pCDNA3-NLbh (Addgene) and the shRNAs. Two days after transfection, the supernatant that contained the virus particles was collected and used for infection of HEK293T cells in the presence of polybrene (8 mg/mL). Continuously expressing SMUG1 shRNA stable cell line was generated in HEK293T cells.
2.5. T7E1 Assay
We performed a T7E1 assay to validate the gene modification in SMUG1 KO cell lines. After we extracted the genomic DNA from SMUG1 KO cell lines, we amplified the CRIPSR/Cas9 target sequence in the SMUG1 gene using target sequence-specific primer (F: 5′ TCC-ATC-CAT-GAG-CCT-GCA-GGT-GCC-C 3′, R: 5′ AAG-GAT-GGG-GCT-GGC-GGA-AGG-AAG-A 3′). The amplified products were denatured at 95 °C for 5 min and annealed in order to allow the formation of a heteroduplex between amplified products with and without mutations. After annealing, the products reacted with the T7E1 enzyme at 37 °C for 15 min. We stopped the reaction by adding 1 µL of 0.5 M EDTA and detected the cleaved mismatch products by using agarose gel electrophoresis.
2.6. Wound Healing Assay
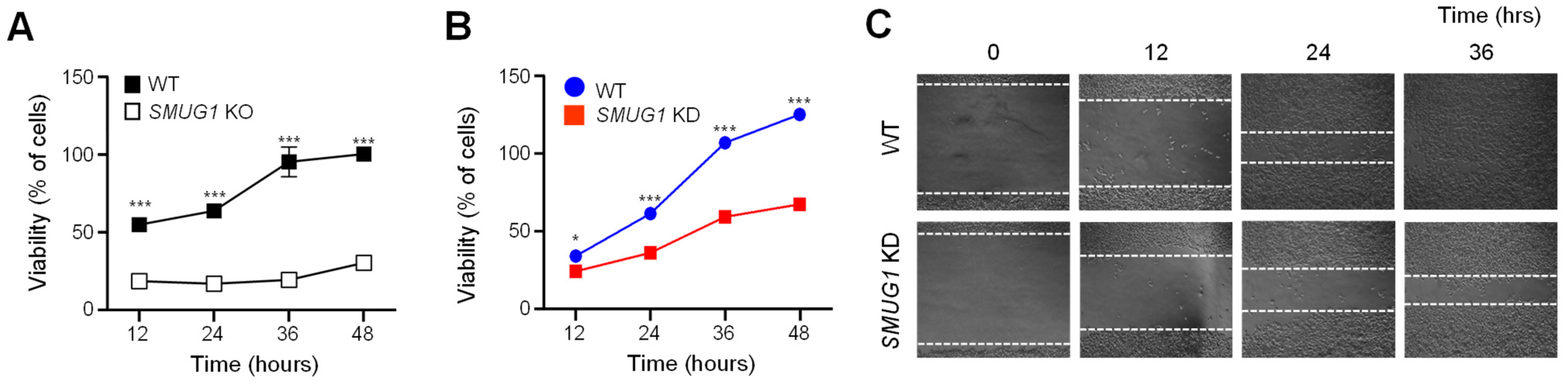
To determine the effects of cell migration, we seeded approximately 6 × 105 cells/well in 24-well culture plates. A straight scratch that simulated a wound was generated by a 100p pipette tip. After scratching, we removed the suspended cells in the medium and washed them with 1 × PBS twice. The migration of both WT and KD cells were photographed at each indicated time.
2.7. RNA-Seq Library Preparation and Sequencing
The RNA-seq library construction was performed using SENSE 3′ mRNA-Seq Library Preparation Kit (Lexogen Inc., Vienna, Austria) according to the manufacturer’s instructions. In brief, each 100 ng total RNA from the cells was prepared, an oligo-dT primer containing an Illumina-compatible sequence at its 5′ end was hybridized to the RNA, and reverse transcription was performed. After degradation of the RNA template, second-strand synthesis was initiated by a random primer containing an Illumina’s compatible linker sequence at its 5′ end. The double-stranded library was purified by using magnetic beads to remove all reaction components. The library was amplified to add the complete adapter sequences required for cluster generation. The finished library was purified from PCR components. High-throughput sequencing was performed as single-end 75 sequencing using Next Seq 500 (Illumina, Inc., San Diego, CA, USA).
2.8. Bioinformatical Analysis
The entire analysis pipeline of RNA-seq was coded by using R (ver. 3.6) which was controlled by systemPipeR (ver. 1.18.2). Raw sequence reads were trimmed for adaptor sequence and masked for low-quality sequences using systemPipeR. Transcript quantification of RNA-seq reads was performed with GenomicAlignments (ver.1.20.1) by reads aligned to Ensemble v95 Homo Sapiens transcriptome annotation (GRCh.38.95) using Rsubread (ver. 1.24.6). The FPKM values were calculated using the “fpkm” function from DESeq2 (ver. 1.24.0) that processed them using the robust median ratio method, and transcript reads were normalized by the “voom” function from Limma (ver. 3.40.6). To analyze a transcript as differentially expressed, EdgeR (ver. 3.26.7) calculated the results based on the normalized counts from entire sequence alignments. Significantly differentially expressed transcripts having greater than the fold change of raw FPKM value > 2 and p value < 0.01 cases in all experimental comparisons were selected and used for further analysis. Gene annotation was added by the online database using Ensemble biomaRt (ver. 2.40.4), and visualization was performed by using R base code and gplots (ver. 3.0.1.1).
2.9. Statistical Analyses
Values are presented as mean ± SEM of experiments performed in triplicate. Data were analyzed by two-way ANOVA followed by Tukey’s multiple comparison test using GraphPad Prism software version 5.01 (San Diego, CA, USA). Differences between groups were considered to be significant at p < 0.05.
4. Discussion
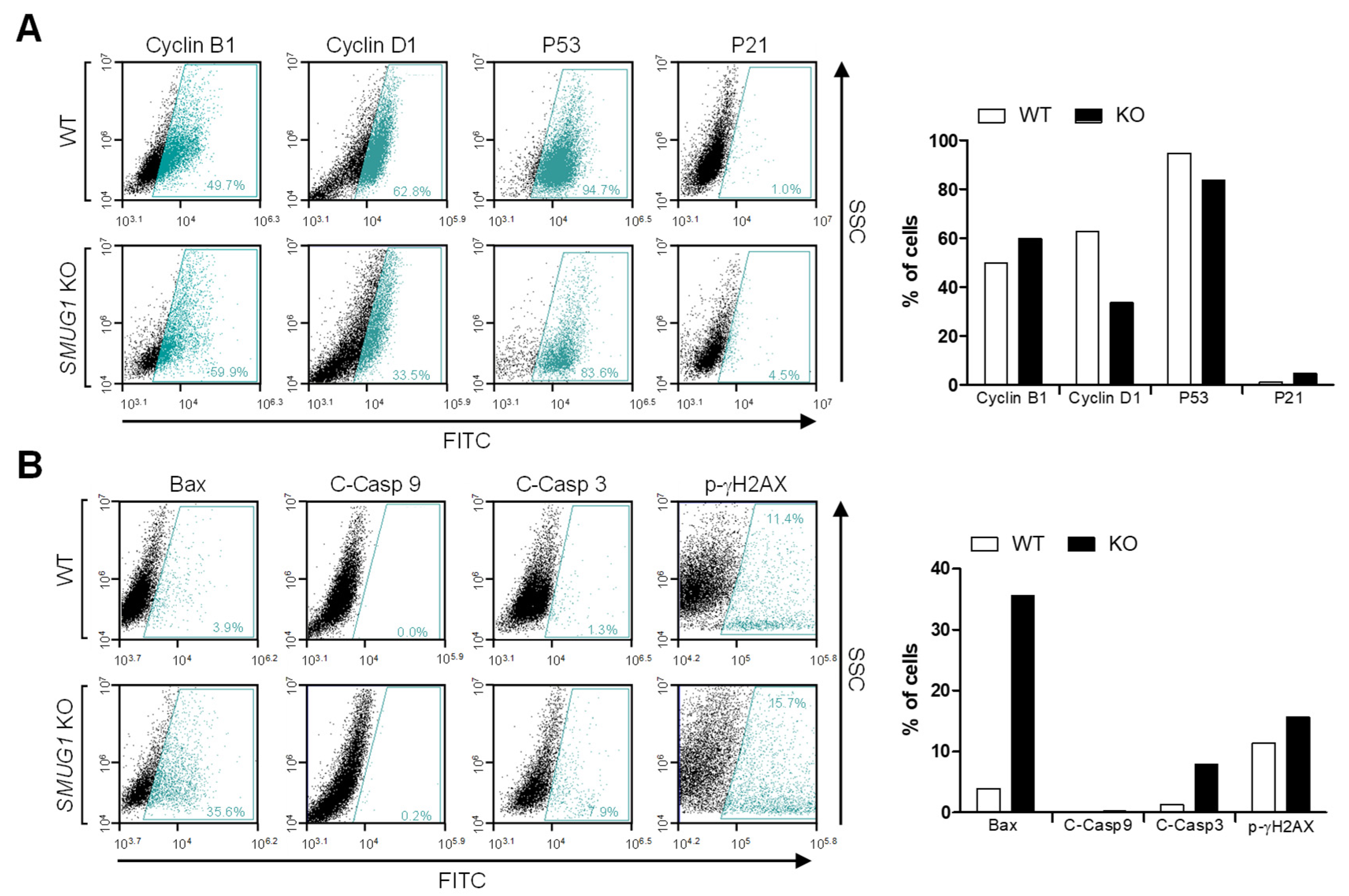
In mammals, a wide range of chemically altered bases was removed through the base excision repair (BER) pathway, and genome integrity was protected by DNA checkpoints, which initiate DNA repair and delay cell cycle progression to prevent replication and segregation of damaged DNA molecules. The transcriptome analysis showed that gene ontology for the regulation of cell cycle (GO:0051726), regulation of cell cycle process (GO:0010564), and regulation of mitotic cell cycle phase transition (GO:19019990) was down-regulated in
SMUG1 KO cells. Among cell cycle regulation genes, the expression of cyclin-dependent kinase 1 (CDK1), which regulates the G2/M transition and mitotic progression, was markedly decreased (
Figure 8F). Moreover, the gene ontology for positive regulation of the intrinsic apoptotic signaling pathway (GO:2001244) and apoptotic mitochondrial changes (GO: 0008637), such as
Beclin 1 (
BECN1), was up-regulated in
SMUG1 KO cells (
Figure 8F). BECN1 interacts with BCL-2, which induces the mitochondrial translocation of BAX and the release of proapoptotic factors. It was shown that SMUG1 regulates gene expression of CDK1 and BECN1, and it inhibits mitotic progression and finally undergoes apoptotic cell death.
The alteration of the cellular genome can produce mutations by base-mispairing during DNA replication or can decrease cell viability. Many previous studies suggest that DNA glycosylases initiate the base excision pathway through the cleavage of the N-glycosidic bond in the DNA backbone, and the biochemical function of DNA glycosylases was essential for repair during DNA replication [
16]. Here, we analyzed the proportion of the cell cycle in
SMUG1 WT and
SMUG1 KO HepG2 cells to better understand the role of
SMUG1 in cell growth and cell division. We observed that the absence of
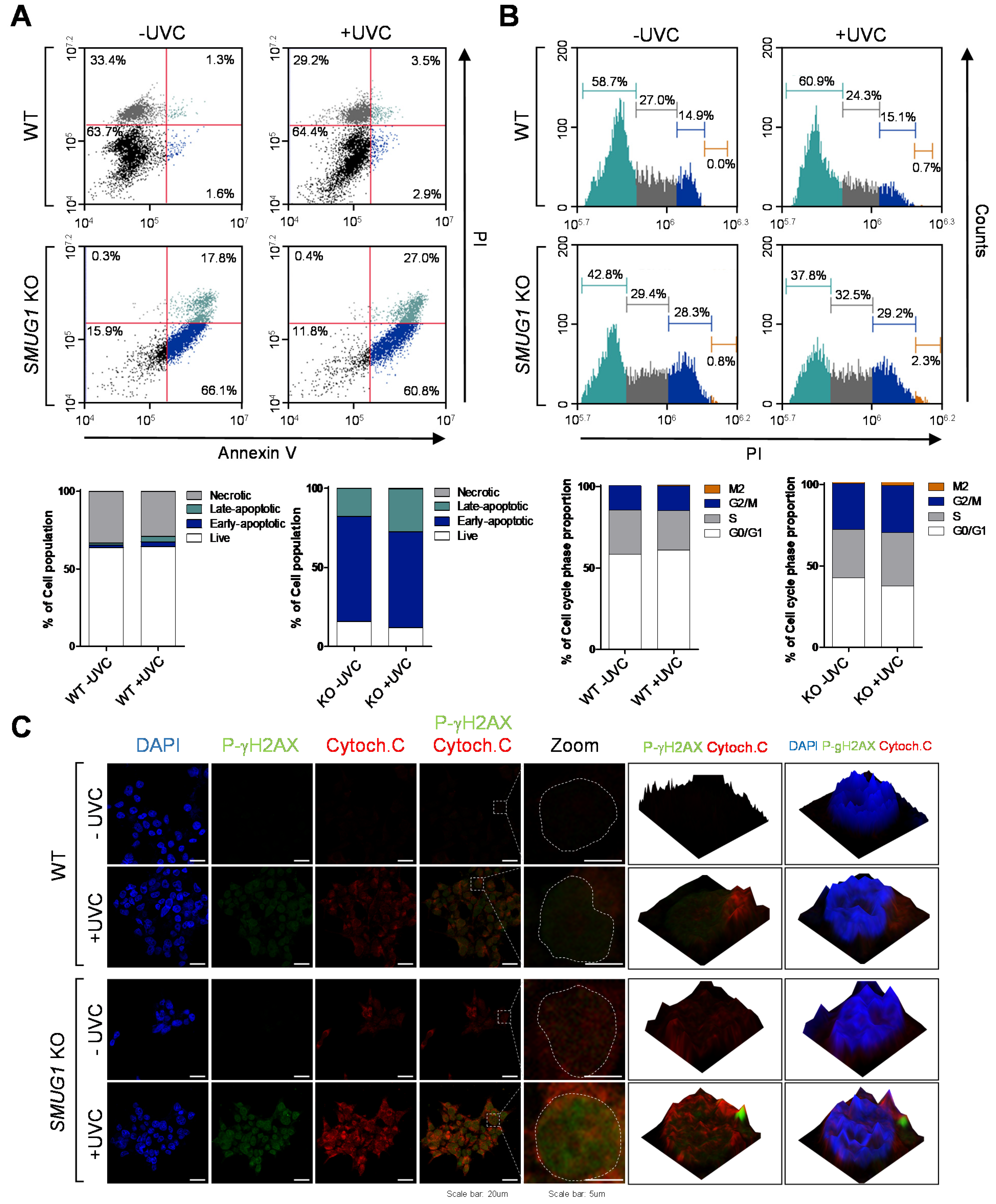
SMUG1 induces accumulation of cells in both S and G2-M phases and that it also promotes apoptosis. Ionizing radiation-induced DNA damage is processed by the BER pathway and BER enzymes throughout the cell cycle to investigate the cell cycle-specific repair [
17]. We have previously shown that the expression of
SMUG1 was reduced in mouse retina after UV irradiation [
13]. In UVC irradiated
SMUG1 KO cells, the cell population in the G2/M phase and undergoing apoptosis is highly increased (
Figure 3). In addition, enrichment of H2AX occurred more frequently in the accessible chromatin in
SMUG1 KO cells. Even though we performed the experiments with various aspects, the exact molecular mechanisms of apoptotic cell death in
SMUG1 KO cells needs to be further elucidated. It may involve the DNA repair or replication regulation pathway. Taken together, deficiency of
SMUG1 induces an unstable initial response to replication stress, and it ultimately promotes apoptotic cell death.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







