
Hunting for Familial Parkinson’s Disease Mutations in the Post Genome Era

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
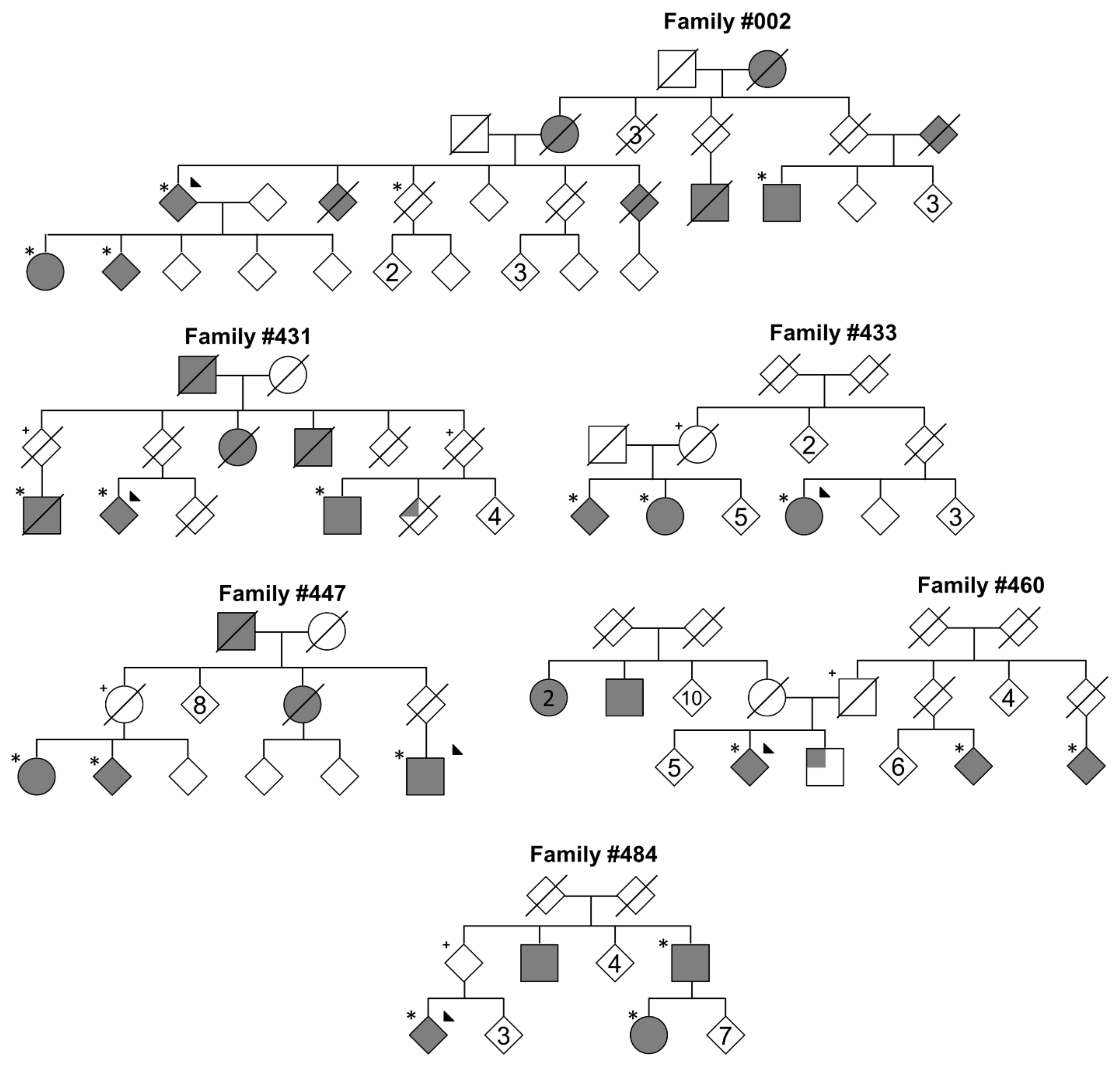
2.1. Patient Recruitment
2.2. Whole Exome Sequencing
2.3. Variant Segregation Analysis
2.4. Mutation Frequency
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bentley, S.R.; Bortnick, S.; Guella, I.; Fowdar, J.Y.; Silburn, P.A.; Wood, S.A.; Farrer, M.J.; Mellick, G.D. Pipeline to gene discovery -Analysing familial Parkinsonism in the Queensland Parkinson’s Project. Parkinsonism Relat. Disord. 2018, 49, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.; Clark, A.J. A pedigree of paralysis agitans. Ann. Eugen. 1926, 1, 455–464. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Trinh, J.; Guella, I.; Farrer, M.J. Disease penetrance of late-onset parkinsonism: A meta-analysis. JAMA Neurol. 2014, 71, 1535–1539. [Google Scholar] [CrossRef]
- Lee, A.J.; Wang, Y.; Alcalay, R.N.; Mejia-Santana, H.; Saunders-Pullman, R.; Bressman, S.; Corvol, J.C.; Brice, A.; Lesage, S.; Mangone, G.; et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1432–1438. [Google Scholar] [CrossRef]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Vilarino-Guell, C.; Rajput, A.; Milnerwood, A.J.; Shah, B.; Szu-Tu, C.; Trinh, J.; Yu, I.; Encarnacion, M.; Munsie, L.N.; Tapia, L.; et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. 2014, 23, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Shi, Y.; Yang, Y.; Ahmeti, K.B.; Miller, N.; Huang, C.; Cheng, L.; Zhai, H.; Deng, S.; Nuytemans, K.; et al. Identification of TMEM230 mutations in familial Parkinson’s disease. Nat. Genet. 2016, 48, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: A genome-wide linkage and sequencing study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef]
- Schulte, E.C.; Stahl, I.; Czamara, D.; Ellwanger, D.C.; Eck, S.; Graf, E.; Mollenhauer, B.; Zimprich, A.; Lichtner, P.; Haubenberger, D.; et al. Rare variants in PLXNA4 and Parkinson’s disease. PLoS ONE 2013, 8, e79145. [Google Scholar] [CrossRef]
- Gasser, T.; Muller-Myhsok, B.; Wszolek, Z.K.; Oehlmann, R.; Calne, D.B.; Bonifati, V.; Bereznai, B.; Fabrizio, E.; Vieregge, P.; Horstmann, R.D. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat. Genet. 1998, 18, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funayama, M.; Hasegawa, K.; Kowa, H.; Saito, M.; Tsuji, S.; Obata, F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann. Neurol. 2002, 51, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Gelb, D.J.; Oliver, E.; Gilman, S. Diagnostic criteria for Parkinson disease. Arch. Neurol. 1999, 56, 33–39. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Makarious, M.B.; Bandres-Ciga, S.; Gibbs, J.R.; Ding, J.; Hernandez, D.G.; Brooks, J.; Grenn, F.P.; Iwaki, H.; Singleton, A.B.; et al. The Parkinson’s Disease DNA Variant Browser. Mov. Disord. Off. J. Mov. Disord. Soc. 2021. [Google Scholar] [CrossRef]
- Pinese, M.; Lacaze, P.; Rath, E.M.; Stone, A.; Brion, M.J.; Ameur, A.; Nagpal, S.; Puttick, C.; Husson, S.; Degrave, D.; et al. The Medical Genome Reference Bank contains whole genome and phenotype data of 2570 healthy elderly. Nat. Commun. 2020, 11, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hout, C.V.; Tachmazidou, I.; Backman, J.D.; Hoffman, J.D.; Liu, D.; Pandey, A.K.; Gonzaga-Jauregui, C.; Khalid, S.; Ye, B.; Banerjee, N.; et al. Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 2020, 586, 749–756. [Google Scholar] [CrossRef]
- Spilker, C.; Kreutz, M.R. RapGAPs in brain: Multipurpose players in neuronal Rap signalling. Eur. J. Neurosci. 2010, 32, 1–9. [Google Scholar] [CrossRef]
- Lu, X.J.; Chen, X.Q.; Weng, J.; Zhang, H.Y.; Pak, D.T.; Luo, J.H.; Du, J.Z. Hippocampal spine-associated Rap-specific GTPase-activating protein induces enhancement of learning and memory in postnatally hypoxia-exposed mice. Neuroscience 2009, 162, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, K.; Zhu, K.; Sun, Y.H.; Hegyi, B.; Zeng, Q.; Murphy, C.J.; Small, J.V.; Chen-Izu, Y.; Izumiya, Y.; Penninger, J.M.; et al. KCNJ15/Kir4.2 couples with polyamines to sense weak extracellular electric fields in galvanotaxis. Nat. Commun. 2015, 6, 8532. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics: Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chen, Y.W.; Tsai, S.F.; Wu, S.N.; Shih, Y.H.; Jiang-Shieh, Y.F.; Yang, T.T.; Kuo, Y.M. Estrogen ameliorates microglial activation by inhibiting the Kir2.1 inward-rectifier K(+) channel. Sci. Rep. 2016, 6, 22864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lucente, J.; Nguyen, H.M.; Wulff, H.; Jin, L.W.; Maezawa, I. The voltage-gated potassium channel Kv1.3 is required for microglial pro-inflammatory activation in vivo. Glia 2018, 66, 1881–1895. [Google Scholar] [CrossRef]
- Klein, C.; Chuang, R.; Marras, C.; Lang, A.E. The curious case of phenocopies in families with genetic Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Nandhagopal, R.; Mak, E.; Schulzer, M.; McKenzie, J.; McCormick, S.; Sossi, V.; Ruth, T.J.; Strongosky, A.; Farrer, M.J.; Wszolek, Z.K.; et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: A multitracer PET study. Neurology 2008, 71, 1790–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Luth, T.; Konig, I.R.; Grunewald, A.; Kasten, M.; Klein, C.; Hentati, F.; Farrer, M.; Trinh, J. Age at Onset of LRRK2 p.Gly2019Ser Is Related to Environmental and Lifestyle Factors. Mov. Disord. Off. J. Mov. Disord. Soc. 2020, 35, 1854–1858. [Google Scholar] [CrossRef]
- Quadri, M.; Yang, X.; Cossu, G.; Olgiati, S.; Saddi, V.M.; Breedveld, G.J.; Ouyang, L.; Hu, J.; Xu, N.; Graafland, J.; et al. An exome study of Parkinson’s disease in Sardinia, a Mediterranean genetic isolate. Neurogenetics 2015, 16, 55–64. [Google Scholar] [CrossRef]
- Kumar, S.; Yadav, N.; Pandey, S.; Muthane, U.B.; Govindappa, S.T.; Abbas, M.M.; Behari, M.; Thelma, B.K. Novel and reported variants in Parkinson’s disease genes confer high disease burden among Indians. Parkinsonism Relat. Disord. 2020, 78, 46–52. [Google Scholar] [CrossRef]
- Siitonen, A.; Nalls, M.A.; Hernandez, D.; Gibbs, J.R.; Ding, J.; Ylikotila, P.; Edsall, C.; Singleton, A.; Majamaa, K. Genetics of early-onset Parkinson’s disease in Finland: Exome sequencing and genome-wide association study. Neurobiol. Aging 2017, 53, 195.e7–195.e10. [Google Scholar] [CrossRef] [Green Version]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; International Parkinson’s Disease Genomics Consortium. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Gaare, J.J.; Skeie, G.O.; Tzoulis, C.; Larsen, J.P.; Tysnes, O.B. Familial aggregation of Parkinson’s disease may affect progression of motor symptoms and dementia. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kun-Rodrigues, C.; Ganos, C.; Guerreiro, R.; Schneider, S.A.; Schulte, C.; Lesage, S.; Darwent, L.; Holmans, P.; Singleton, A.; International Parkinson’s Disease Genomics Consortium; et al. A systematic screening to identify de novo mutations causing sporadic early-onset Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 6711–6720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Ye, H.; Heetveld, S.; Lechler, M.C.; Michels, H.; Seinstra, R.I.; Lubbe, S.J.; Drouet, V.; Lesage, S.; Majounie, E.; et al. Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol. 2017, 18, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Family | # of Affected Carriers | # of Unaffected Carriers I | Gene | Transcript | Protein Change | GnomAD Exome MAF | AnnEx State (Count) | PD Genetics | UKB II and MGRB | CADD (Phred) |
|---|---|---|---|---|---|---|---|---|---|---|
| #002 | 5 | 2 | KCNJ15 | NM_001276436 | p.R28C | 1.22 × 10−5 | Non-PD (1) | 0 | 4 | 16.9 |
| SON | NM_138927 | p.S1595P | 4.06 × 10−5 | 0 | 0 | 13 | 2.864 | |||
| 3 | PASK | NM_001252120 | p.P519L | 2.03 × 10−5 | 0 | 2 | 3 | 11.58 | ||
| #431 | 3 | 2 | ARL14EP | NM_152316 | p.A146V | 0.00 | 0 | 0 | 0 | 21.5 |
| HYDIN | NM_001270974 | p.A2271E | 4.44 × 10−6 | 0 | 0 | 0 | 4.081 | |||
| PTPRA | NM_080840 | p.R223W | 5.32 × 10−5 | Non-PD (2) | 0 | 43 | 28.6 | |||
| #433 | 3 | 1 | SIPA1L1 | NM_001284247 | p.R236Q | 2.85 × 10−5 | 0 | 0 | 13 | 16.02 |
| #447 | 3 | 1 | ZNF462 | NM_021224 | p.I1523V | 4.06 × 10−6 | 0 | 0 | 0 | 11.9 |
| DUSP19 | NM_001142314 | p.I111R | 4.06 × 10−6 | 0 | 0 | 0 | 24 | |||
| KCTD1 | NM_001136205 | p.G134R | 4.07 × 10−6 | 0 | 0 | 0 | 4.792 | |||
| TAF1C | NM_001243158 | p.R346Q | 8.29 × 10−6 | 0 | 1 | 0 | 2.672 | |||
| DARS2 | NM_018122 | p.S59L | 1.22 × 10−5 | 0 | 0 | 0 | 15.29 | |||
| EXPH5 | NM_001144765 | p.T920S | 2.88 × 10−5 | 0 | 0 | 1 | 10.15 | |||
| FAM71B | NM_130899 | p.I318T | 6.10 × 10−5 | Non-PD (2) III | 1 | 2 | 0.01 | |||
| CCDC180 | NM_020893 | p.R1684C | 8.53 × 10−5 | 0 | 0 | 3 | 12.13 | |||
| #484 | 3 | 1 | TCP10L | NM_144659 | p.I54F | 0.00 | 0 | 0 | 0 | 5.434 |
| ADCY4 | NM_001198568 | p.H760Q | 4.10 × 10−6 | 0 | 0 | 6 | 11.05 | |||
| USP42 | NM_032172 | p.H952R | 5.69 × 10−6 | 0 | 0 | 1 | 14.75 | |||
| UNC13B | NM_006377 | c.3188+1G>A | 1.63 × 10−5 | 0 | 1 | 0 | 25.6 | |||
| CFTR | NM_000492 | p.K1080R | 2.85 × 10−5 | Non-PD (2) | 0 | 1 | 28.7 | |||
| NINL | NM_025176 | p.Q1232H | 3.58 × 10−5 | 0 | 0 | 9 | 12.17 | |||
| CST5 | NM_001900 | p.S66N | 3.67 × 10−5 | Non-PD (3) | 4 | 12 | 11.77 | |||
| #460 | 3 IV | 1 | SLC2A12 | NM_145176 | p.S357L | 0.00 | 0 | 0 | 0 | 22 |
| DOCK3 | NM_004947 | p.R392W | 0.00 | 0 | 0 | 0 | 18.96 | |||
| TPR | NM_003292 | p.K1038N | 0.00 | 0 | 0 | 4 | 16.34 | |||
| DNAH1 | NM_015512 | p.K1792R | 4.06 × 10−6 | 0 | 0 | 0 | 21.3 | |||
| PCDHGA7 | NM_018920 | p.S667G | 1.63 × 10−5 | Non-PD (1) | 1 | 12 | 12.13 | |||
| MYOT | NM_001135940 | p.N30K | 2.03 × 10−5 | Non-PD (1) | 1 | 10 | 6.21 | |||
| KIF9 | NM_022342 | p.R287W | 2.44 × 10−5 | 0 | 0 | 0 | 20.4 | |||
| DNAJC12 | NM_021800 | p.T99M | 4.06 × 10−5 | Non-PD (1) | 2 | 6 | 14.86 | |||
| SLAMF8 | NM_020125 | p.V234E | 7.31 × 10−5 | Non-PD (1) | 2 | 21 | 17.11 | |||
| ZNF75A | NM_153028 | p.Q212E | 8.95 × 10−5 | Non-PD (1), PD(2) V | 0 | 28 | 14.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bentley, S.R.; Guella, I.; Sherman, H.E.; Neuendorf, H.M.; Sykes, A.M.; Fowdar, J.Y.; Silburn, P.A.; Wood, S.A.; Farrer, M.J.; Mellick, G.D. Hunting for Familial Parkinson’s Disease Mutations in the Post Genome Era. Genes 2021, 12, 430. https://doi.org/10.3390/genes12030430
Bentley SR, Guella I, Sherman HE, Neuendorf HM, Sykes AM, Fowdar JY, Silburn PA, Wood SA, Farrer MJ, Mellick GD. Hunting for Familial Parkinson’s Disease Mutations in the Post Genome Era. Genes. 2021; 12(3):430. https://doi.org/10.3390/genes12030430
Chicago/Turabian StyleBentley, Steven R., Ilaria Guella, Holly E. Sherman, Hannah M. Neuendorf, Alex M. Sykes, Javed Y. Fowdar, Peter A. Silburn, Stephen A. Wood, Matthew J. Farrer, and George D. Mellick. 2021. "Hunting for Familial Parkinson’s Disease Mutations in the Post Genome Era" Genes 12, no. 3: 430. https://doi.org/10.3390/genes12030430
APA StyleBentley, S. R., Guella, I., Sherman, H. E., Neuendorf, H. M., Sykes, A. M., Fowdar, J. Y., Silburn, P. A., Wood, S. A., Farrer, M. J., & Mellick, G. D. (2021). Hunting for Familial Parkinson’s Disease Mutations in the Post Genome Era. Genes, 12(3), 430. https://doi.org/10.3390/genes12030430