
Solid Pseudopapillary Neoplasm of the Pancreas and Abdominal Desmoid Tumor in a Patient Carrying Two Different BRCA2 Germline Mutations: New Horizons from Tumor Molecular Profiling

 , , , ,
, , , , 
 , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
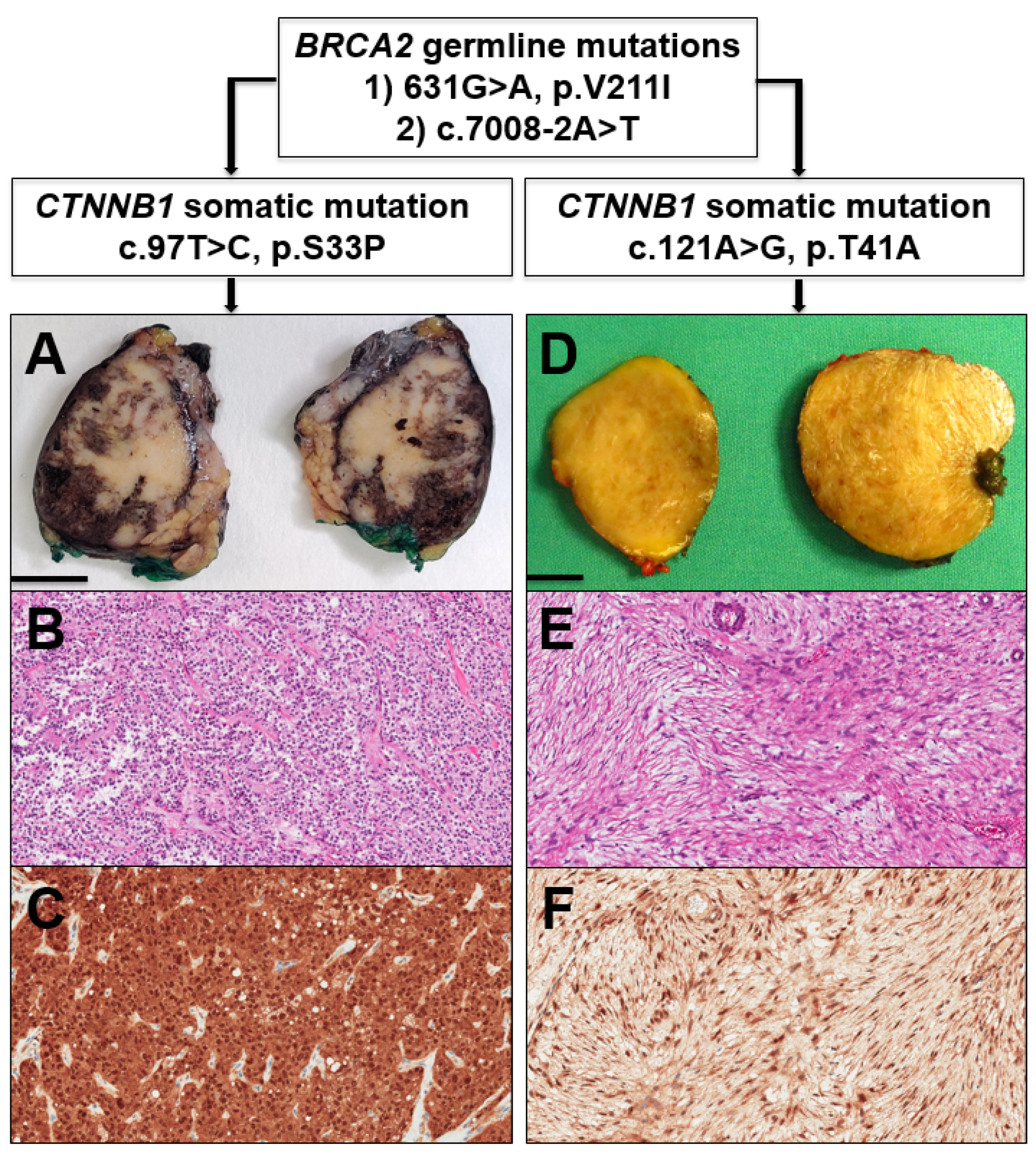
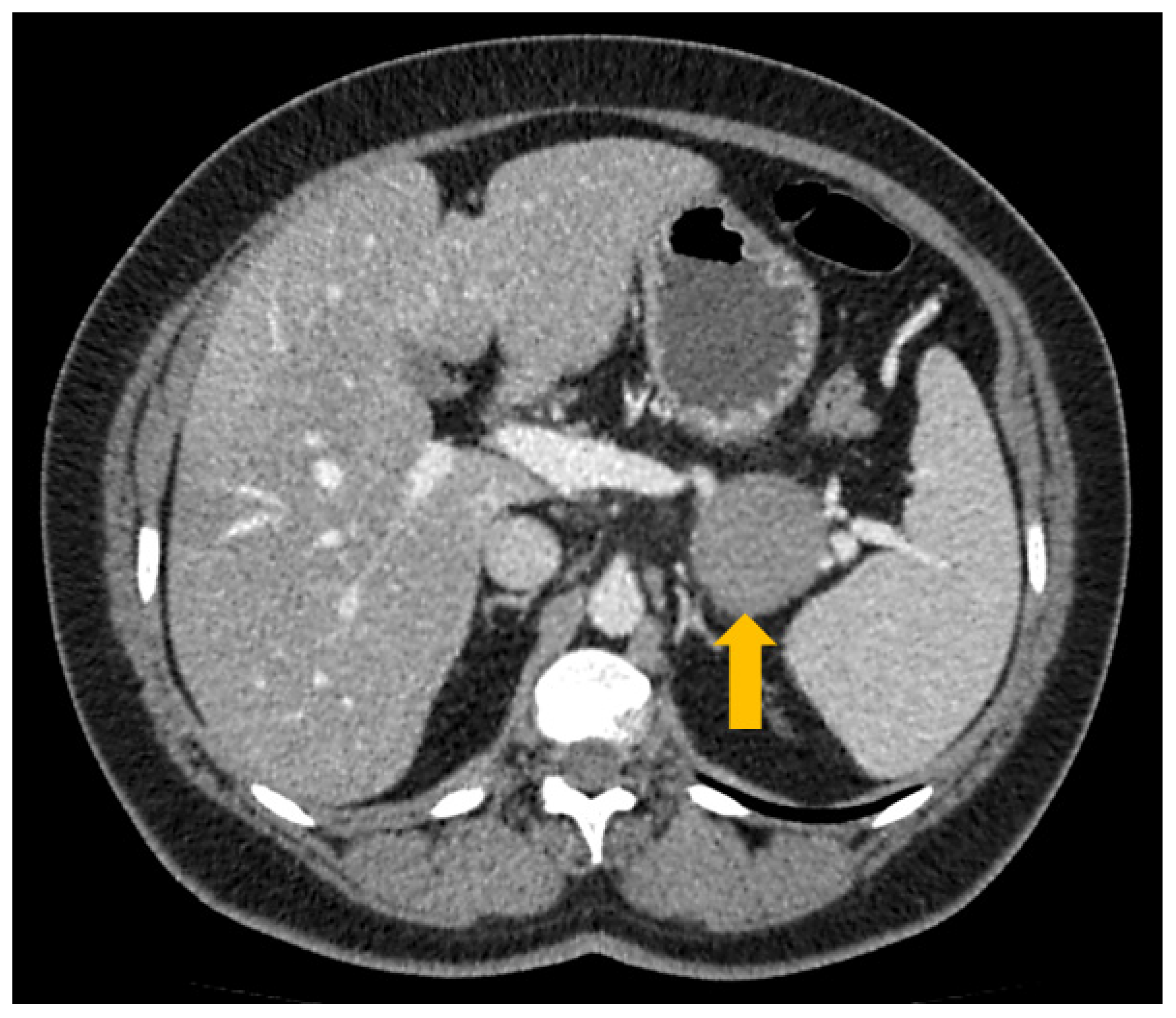
2. Case Report and Discussion
3. Materials and Methods
3.1. Immunohistochemistry
3.2. Molecular Analysis
3.2.1. DNA Extraction
3.2.2. Massive Parallel Sequencing (Next-Generation Sequencing, NGS)
3.2.3. Variant Classification
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- International Agency for Research on Cancer. WHO Classification of Tumours Editorial Board. Digestive System Tumors, 5th ed.; IARC Press: Lyon, France, 2019.
- Marchegiani, G.; Andrianello, S.; Massignani, M.; Malleo, G.; Maggino, L.; Paiella, S.; Ferrone, C.R.; Luchini, C.; Scarpa, A.; Capelli, P.; et al. Solid pseudopapillary tumors of the pancreas: Specific pathological features predict the likelihood of postoperative recurrence. J. Surg. Oncol. 2016, 114, 597–601. [Google Scholar] [CrossRef]
- Lee, G.; Sung, Y.N.; Kim, S.J.; Lee, J.H.; Song, K.B.; Hwang, D.W.; Kim, J.; Lee, S.S.; Kim, S.C.; Hong, S.M. Large tumor size, lymphovascular invasion, and synchronous metastasis are associated with the recurrence of solid pseudopapillary neoplasms of the pancreas. HPB 2021, 23, 220–230. [Google Scholar] [CrossRef]
- Wu, J.; Jiao, Y.; Dal Molin, M.; Maitra, A.; de Wilde, R.F.; Wood, L.D.; Eshleman, J.R.; Goggins, M.G.; Wolfgang, C.L.; Canto, M.I.; et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 21188–21193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Kim, M.; Hwang, D.; Park, M.; Kim, W.K.; Kim, S.K.; Shin, J.; Park, E.S.; Kang, C.M.; Paik, Y.K.; et al. Characterization of gene expression and activated signaling pathways in solid-pseudopapillary neoplasm of pancreas. Mod. Pathol. 2014, 27, 580–593. [Google Scholar] [CrossRef] [Green Version]
- Amato, E.; Mafficini, A.; Hirabayashi, K.; Lawlor, R.T.; Fassan, M.; Vicentini, C.; Barbi, S.; Delfino, P.; Sikora, K.; Rusev, B.; et al. Molecular alterations associated with metastases of solid pseudopapillary neoplasms of the pancreas. J. Pathol. 2019, 247, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.; Jeong, S. Multitasking β-catenin: From adhesion and transcription to RNA regulation. Anim. Cells Syst. 2013, 17, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Bashyam, M.D. Multiple oncogenic roles of nuclear β-catenin. J. Biosci. 2017, 42, 695–707. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Kasper, B.; Raut, C.P.; Gronchi, A. Desmoid tumors: To treat or not to treat, that is the question. Cancer 2020, 126, 5213–5221. [Google Scholar] [CrossRef]
- Penel, N.; Chibon, F.; Salas, S. Adult desmoid tumors: Biology, management and ongoing trials. Curr. Opin. Oncol. 2017, 29, 268–274. [Google Scholar] [CrossRef]
- De Marchis, M.L.; Tonelli, F.; Quaresmini, D.; Lovero, D.; Della-Morte, D.; Silvestris, F.; Guadagni, F.; Palmirotta, R. Desmoid Tumors in Familial Adenomatous Polyposis. Anticancer Res. 2017, 37, 3357–3366. [Google Scholar]
- Mafficini, A.; Simbolo, M.; Parisi, A.; Rusev, B.; Luchini, C.; Cataldo, I.; Piazzola, E.; Sperandio, N.; Turri, G.; Franchi, M.; et al. BRCA somatic and germline mutation detection in paraffin embedded ovarian cancers by next-generation sequencing. Oncotarget 2016, 7, 1076–1083. [Google Scholar] [CrossRef] [Green Version]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Mody, R.J.; Wu, Y.M.; Lonigro, R.J.; Cao, X.; Roychowdhury, S.; Vats, P.; Frank, K.M.; Prensner, J.R.; Asangani, I.; Palanisamy, N.; et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA 2015, 314, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fabre, M.; Branchereau, S.; Gauthier, F.; Perilongo, G.; Buendia, M.A. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene 2000, 19, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Denkhaus, D.; Albrecht, S.; Leuschner, I.; von Schweinitz, D.; Pietsch, T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res. 1999, 59, 269–273. [Google Scholar] [PubMed]
- Legoix, P.; Bluteau, O.; Bayer, J.; Perret, C.; Balabaud, C.; Belghiti, J.; Franco, D.; Thomas, G.; Laurent-Puig, P.; Zucman-Rossi, J. Beta-catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene 1999, 18, 4044–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitoh, K.; Konishi, F.; Iijima, T.; Ohdaira, T.; Sakai, K.; Kanazawa, K.; Miyaki, M. A novel case of a sporadic desmoid tumour with mutation of the beta catenin gene. J. Clin. Pathol 1999, 52, 695–696. [Google Scholar] [CrossRef] [Green Version]
- Mucaki, E.J.; Ainsworth, P.; Rogan, P.K. Comprehensive prediction of mRNA splicing effects of BRCA1 and BRCA2 variants. Hum. Mutat. 2011, 32, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; De Vecchi, G.; Caleca, L.; Foglia, C.; Ripamonti, C.B.; Ficarazzi, F.; Barile, M.; Varesco, L.; Peissel, B.; Manoukian, S.; et al. Comparative in vitro and in silico analyses of variants in splicing regions of BRCA1 and BRCA2 genes and characterization of novel pathogenic mutations. PLoS ONE 2013, 8, e57173. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2016, 34, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Dubbink, H.J.; Hollink, I.H.I.M.; Avenca Valente, C.; Wang, W.; Liu, P.; Doukas, M.; van Noesel, M.M.; Dinjens, W.N.M.; Wagner, A.; Smits, R. A novel tissue-based ß-catenin gene and immunohistochemical analysis to exclude familial adenomatous polyposis among children with hepatoblastoma tumors. Pediatr. Blood Cancer 2018, 65, e26991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchini, C.; Lawlor, R.T.; Milella, M.; Scarpa, A. Molecular Tumor Boards in Clinical Practice. Trends Cancer 2020, 6, 738–744. [Google Scholar] [CrossRef]
- Nelson, H.D.; Pappas, M.; Cantor, A.; Haney, E.; Holmes, R. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer in Women: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2019, 322, 666–685. [Google Scholar] [CrossRef] [Green Version]
- Sabiani, L.; Barrou, J.; Mathis, J.; Eisinger, F.; Bannier, M.; Lambaudie, E.; Houvenaeghel, G. How to manage BRCA mutation carriers? Horm Mol. Biol. Clin. Investig. 2020, 41. [Google Scholar] [CrossRef]
- Luchini, C.; Parcesepe, P.; Nottegar, A.; Parolini, C.; Mafficini, A.; Remo, A.; Chilosi, M.; Manfrin, E. CD71 in Gestational Pathology: A Versatile Immunohistochemical Marker With New Possible Applications. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 215–220. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Daprà, V.; Girolami, I.; Pea, A.; Pilati, C.; Nottegar, A.; Piccoli, P.; Parolini, C.; Sperandio, N.; Capelli, P.; et al. CD200 expression is a feature of solid pseudopapillary neoplasms of the pancreas. Virchows Arch. 2019, 474, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Simbolo, M.; Gottardi, M.; Corbo, V.; Fassan, M.; Mafficini, A.; Malpeli, G.; Lawlor, R.T.; Scarpa, A. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS ONE 2013, 8, e62692. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischler, G.; Leonard, S. biobambam: Tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 2014, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstung, M.; Papaemmanuil, E.; Campbell, P.J. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics 2014, 30, 1198–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Patel, V.M.; Coon, M.; Nguyen, T.; Land, S.J.; Ruden, D.M.; Lu, X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front. Genet. 2012, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Pritchard, B.; Rios, D.; Chen, Y.; Flicek, P.; Cunningham, F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26, 2069–2070. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Papke, D.J.; Nowak, J.A.; Yurgelun, M.B.; Frieden, A.; Srivastava, A.; Lindeman, N.I.; Sholl, L.M.; MacConaill, L.E.; Dong, F. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod. Pathol. 2018, 31, 1882–1890. [Google Scholar] [CrossRef]
- Ahdesmäki, M.J.; Chapman, B.A.; Cingolani, P.; Hofmann, O.; Sidoruk, A.; Lai, Z.; Zakharov, G.; Rodichenko, M.; Alperovich, M.; Jenkins, D.; et al. Prioritisation of structural variant calls in cancer genomes. PeerJ 2017, 5, e3166. [Google Scholar] [CrossRef] [Green Version]
- Gundem, G.; Perez-Llamas, C.; Jene-Sanz, A.; Kedzierska, A.; Islam, A.; Deu-Pons, J.; Furney, S.J.; Lopez-Bigas, N. IntOGen: Integration and data mining of multidimensional oncogenomic data. Nat. Methods 2010, 7, 92–93. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mafficini, A.; Lawlor, R.T.; Ghimenton, C.; Antonello, D.; Cantù, C.; Paolino, G.; Nottegar, A.; Piredda, M.L.; Salvia, R.; Milella, M.; et al. Solid Pseudopapillary Neoplasm of the Pancreas and Abdominal Desmoid Tumor in a Patient Carrying Two Different BRCA2 Germline Mutations: New Horizons from Tumor Molecular Profiling. Genes 2021, 12, 481. https://doi.org/10.3390/genes12040481
Mafficini A, Lawlor RT, Ghimenton C, Antonello D, Cantù C, Paolino G, Nottegar A, Piredda ML, Salvia R, Milella M, et al. Solid Pseudopapillary Neoplasm of the Pancreas and Abdominal Desmoid Tumor in a Patient Carrying Two Different BRCA2 Germline Mutations: New Horizons from Tumor Molecular Profiling. Genes. 2021; 12(4):481. https://doi.org/10.3390/genes12040481
Chicago/Turabian StyleMafficini, Andrea, Rita T. Lawlor, Claudio Ghimenton, Davide Antonello, Cinzia Cantù, Gaetano Paolino, Alessia Nottegar, Maria L. Piredda, Roberto Salvia, Michele Milella, and et al. 2021. "Solid Pseudopapillary Neoplasm of the Pancreas and Abdominal Desmoid Tumor in a Patient Carrying Two Different BRCA2 Germline Mutations: New Horizons from Tumor Molecular Profiling" Genes 12, no. 4: 481. https://doi.org/10.3390/genes12040481
APA StyleMafficini, A., Lawlor, R. T., Ghimenton, C., Antonello, D., Cantù, C., Paolino, G., Nottegar, A., Piredda, M. L., Salvia, R., Milella, M., Dei Tos, A. P., Fassan, M., Scarpa, A., & Luchini, C. (2021). Solid Pseudopapillary Neoplasm of the Pancreas and Abdominal Desmoid Tumor in a Patient Carrying Two Different BRCA2 Germline Mutations: New Horizons from Tumor Molecular Profiling. Genes, 12(4), 481. https://doi.org/10.3390/genes12040481