
Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families

 , , and
, , and 
Abstract
:1. Introduction
2. Objectives
- -
- To evaluate the implementation of NGS for mutation screening in congenital cataracts at a tertiary hospital in Spain.
- -
- To identify the mutations that produce congenital cataracts in Spain and compare them with those reported for other populations.
3. Results
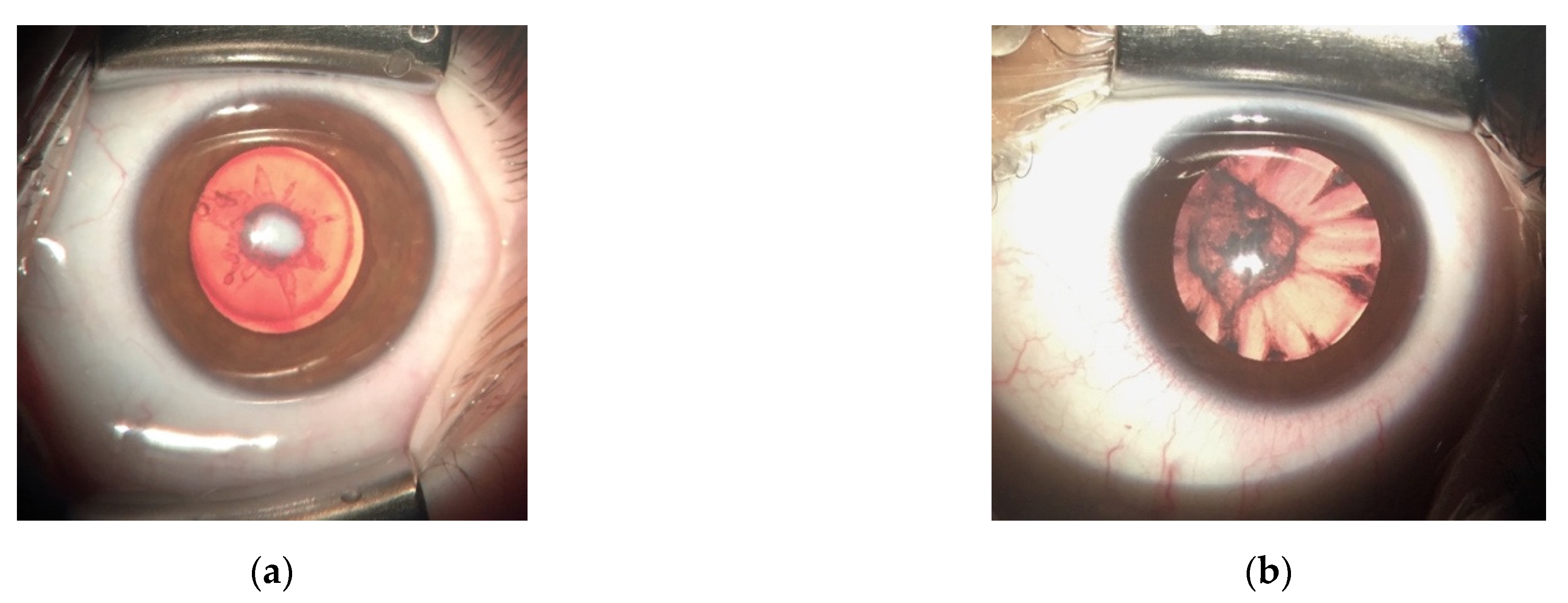
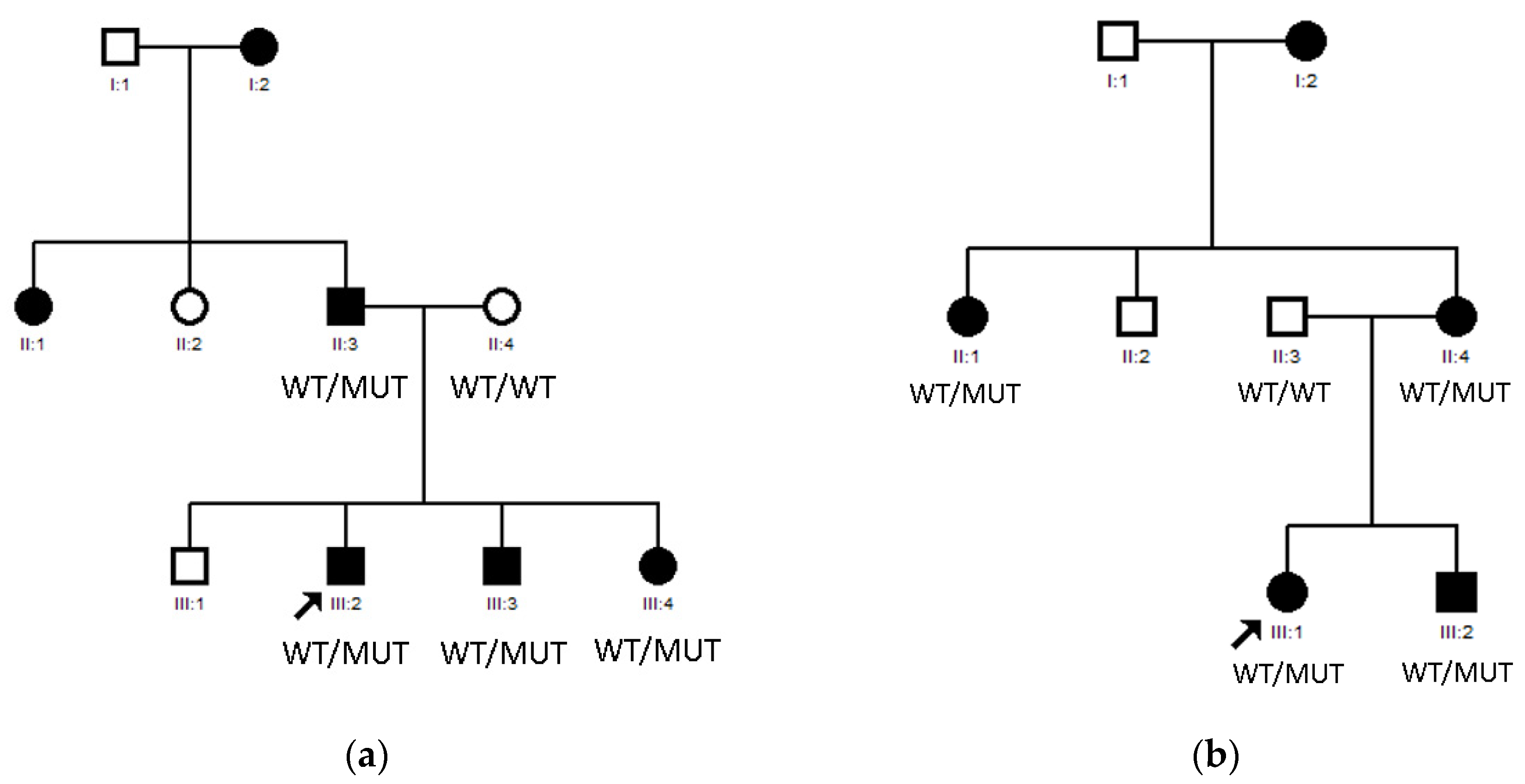
3.1. LIM2 Mutations in Two Spanish Families
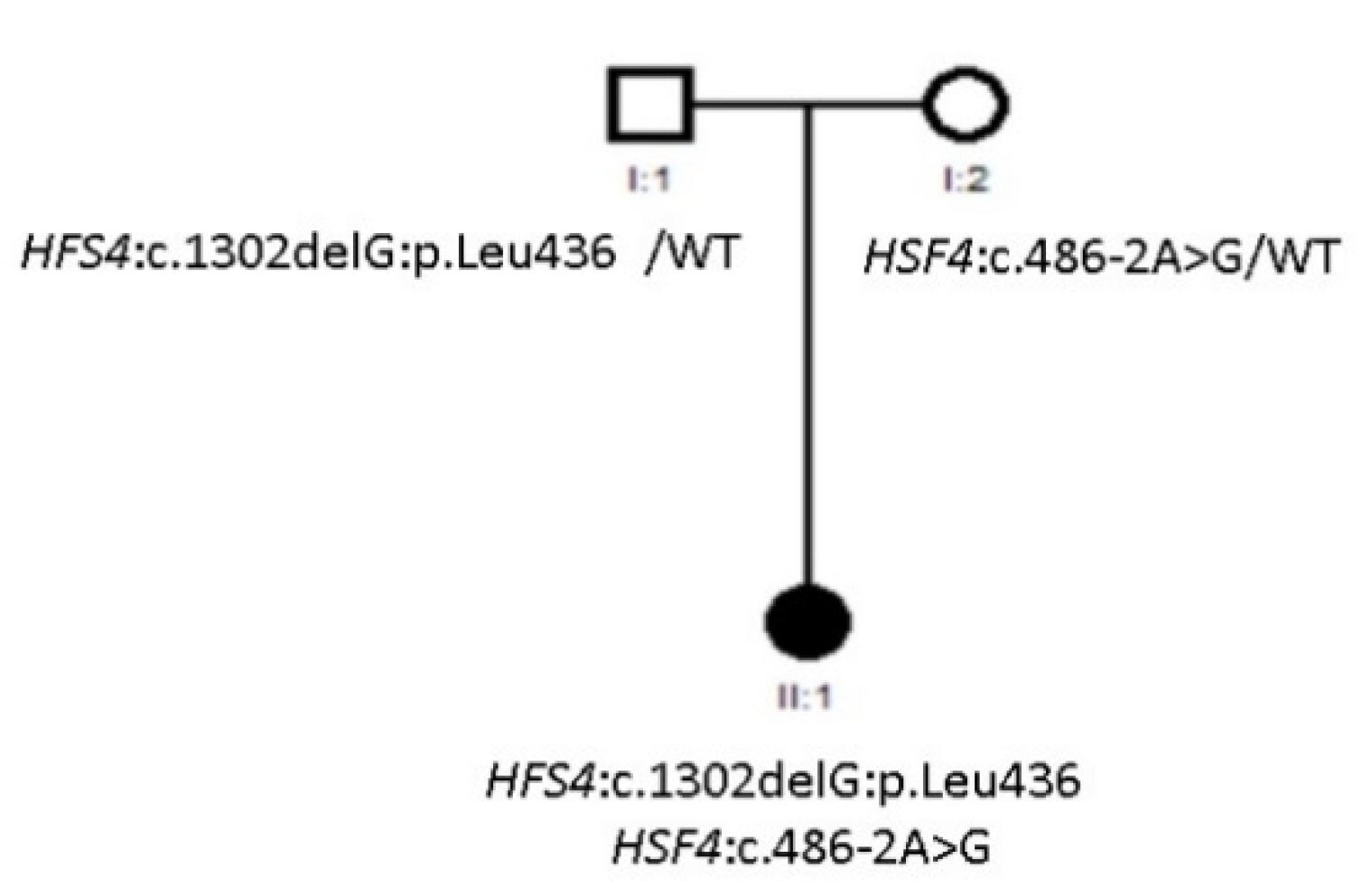
3.2. Compound Heterozygous HSF4 Mutation in a Spanish Family
4. Discussion
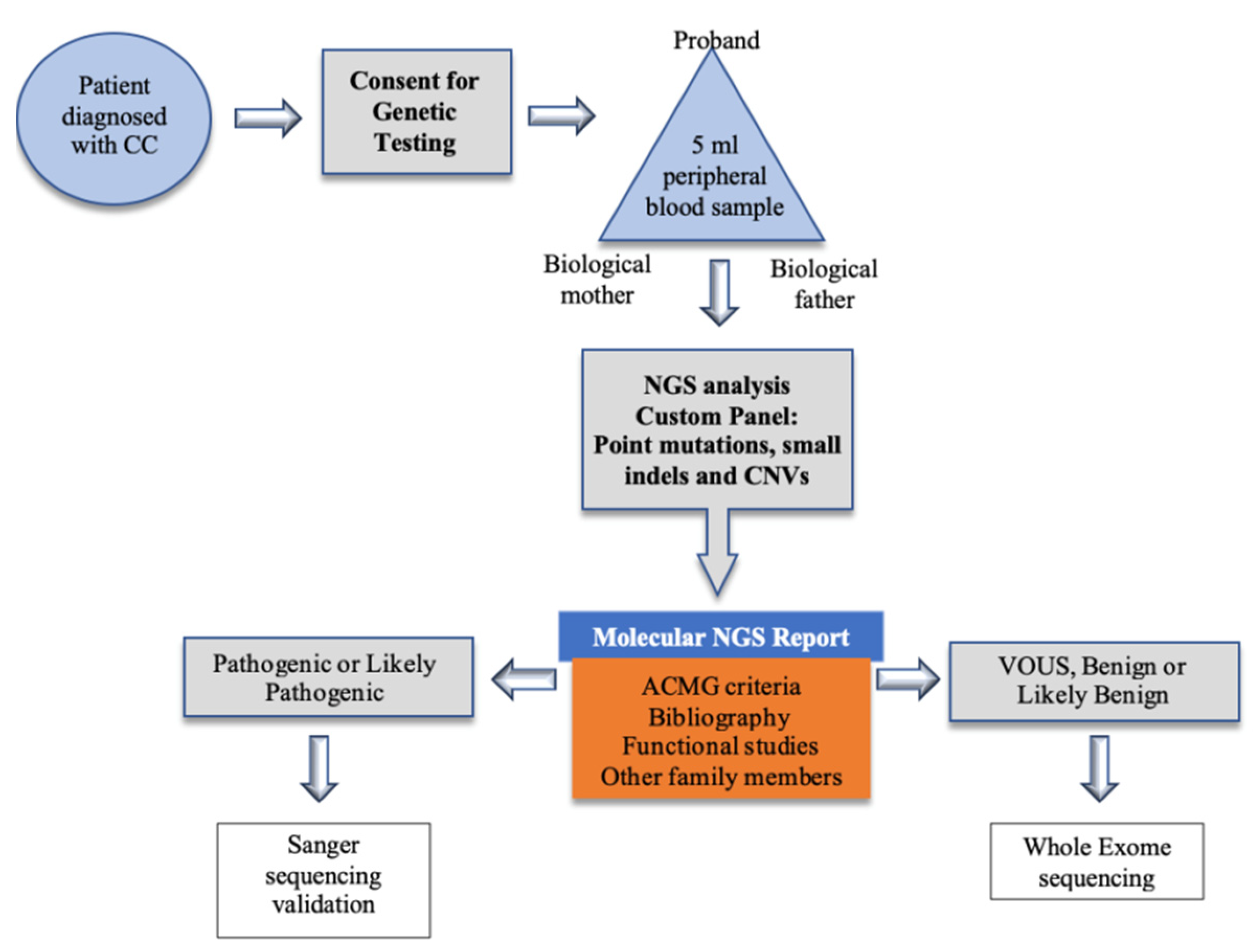
5. Materials and Methods
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bassnet, S.; Jrvoje, S. Lens Growth Process Steven. Physiol. Behav. 2016, 176, 139–148. [Google Scholar]
- Hejtmancik, J.F.; Shiels, A. Overview of the Lens. Prog. Mol. Biol. Transl. Sci. 2015, 134, 119–127. [Google Scholar] [PubMed] [Green Version]
- Lovicu, F.J.; McAvoy, J.W. Growth factor regulation of lens development. Dev. Biol. 2005, 280, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, L.; Shaheen, N.; Hanif, Q.; Fahad, S.; Usman, M. Genetics of congenital cataract, its diagnosis and therapeutics. Egypt J. Basic Appl. Sci. 2018, 5, 252–257. [Google Scholar] [CrossRef]
- Kuszak, J.R.; Zoltoski, R.K.; Sivertson, C. Fibre cell organization in crystalline lenses. Exp. Eye Res. 2004, 78, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; He, W. Molecular characteristics of inherited congenital cataracts. Eur. J. Med. Genet. 2010, 53, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Messina-Baas, O.; Cuevas-Covarrubias, S.A. Inherited Congenital Cataract: A Guide to Suspect the Genetic Etiology in the Cataract Genesis. Mol. Syndromol. 2017, 8, 58–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, C.; Foster, A. Childhood blindness in the context of VISION 2020-The right to sight. Bull. World Health Organ. 2001, 79, 227–232. [Google Scholar]
- Shiels, A.; Hejtmancik, J.F. Mutations and mechanisms in congenital and age-related cataracts. Exp. Eye Res. 2017, 156, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Renwick, J.H.; Lawler, S.D. Probable linkage between a congenital cataract locus and the Duffy blood group locus. Ann. Hum. Genet. 1963, 27, 67–76. [Google Scholar] [CrossRef]
- Bökenkamp, A.; Ludwig, M. The oculocerebrorenal syndrome of Lowe: An update. Pediatr. Nephrol. 2016, 31, 2201–2212. [Google Scholar] [CrossRef] [Green Version]
- Amaya, L.; Taylor, D.; Russell-Eggitt, I.; Nischal, K.K.; Lengyel, D. The morphology and natural history of childhood cataracts. Surv. Ophthalmol. 2003, 48, 125–144. [Google Scholar] [CrossRef]
- Merin, S.; Crawford, J.S. The etiology of congenital cataracts. A survey of 386 cases. Can. J. Ophthalmol. 1971, 6, 178–182. [Google Scholar] [PubMed]
- Lin, H.; Lin, D.; Liu, Z.; Long, E.; Wu, X.; Cao, Q.; Chen, J.; Lin, Z.; Li, X.; Zhang, L.; et al. A novel congenital cataract category system based on lens opacity locations and relevant anterior segment characteristics. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6389–6395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejtmancik, J.F. Congenital Cataracts and their Molecular Genetics. Semin. Cell Dev. Biol. 2008, 19, 134–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, L.M.; Semina, E.V. Genetic landscape of isolated pediatric cataracts: Extreme heterogeneity and variable inheritance patterns within genes. Hum. Genet. 2019, 138, 847–863. [Google Scholar] [CrossRef]
- Alan Shiels, J.F.H. Biology of Cataracts and Opportunities for Treatment. Annu. Rev. Vis. Sci. 2019, 5, 123–149. [Google Scholar] [CrossRef]
- Bhat, S.P. Crystallins, genes and cataract. Prog. Drug Res. 2003, 60, 205–262. [Google Scholar] [PubMed]
- Berry, V.; Georgiou, M.; Fujinami, K.; Quinlan, R.; Moore, A.; Michaelides, M. Inherited cataracts: Molecular genetics, clinical features, disease mechanisms and novel therapeutic approaches. Br. J. Ophthalmol. 2020, 104, 1331–1337. [Google Scholar] [CrossRef] [Green Version]
- Piatigorsky, J. Enigma of the abundant water-soluble cytoplasmic proteins of the cornea: The “refracton” hypothesis. Cornea 2001, 20, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Slingsby, C.; Wistow, G.J. Functions of crystallins in and out of lens: Roles in elongated and post-mitotic cells. Prog. Biophys. Mol. Biol. 2014, 115, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Sinn, R.W. An eye on eye development. J. Mech. Dev. 2013, 130, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.K.H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, L.M.; Semina, E.V. Genetics of anterior segment dysgenesis disorders. Curr. Opin. Ophthalmol. 2011, 22, 314–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants. Acta Ophthalmol. 2018, 96, 134. [Google Scholar]
- Shi, Y.; De Maria, A.B.; Wang, H.; Mathias, R.T.; FitzGerald, P.G.; Bassnett, S. Further analysis of the lens phenotype in Lim2-deficient mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7332–7339. [Google Scholar] [CrossRef] [Green Version]
- Irum, B.; Khan, S.Y.; Ali, M.; Kaul, H.; Kabir, F.; Rauf, B.; Fatima, F.; Nadeem, R.; Khan, A.O.; Al Obaisi, S.; et al. Mutation in LIM2 is responsible for autosomal recessive congenital cataracts. PLoS ONE 2016, 11, e0162620. [Google Scholar] [CrossRef]
- Steele, E.C.; Wang, J.H.; Lo, W.K.; Saperstein, D.A.; Li, X.L.; Church, R.L. Lim2To3 transgenic mice establish a causative relationship between the mutation identified in the Lim2 gene and cataractogenesis in the To3 mouse mutant. Mol. Vis. 2000, 6, 85–94. [Google Scholar]
- Pei, R.; Liang, P.F.; Ye, W.; Li, J.; Ma, J.Y.; Zhou, J. A novel mutation of LIM2 causes autosomal dominant membranous cataract in a Chinese family. Int. J. Ophthalmol. 2020, 13, 1512–1520. [Google Scholar] [CrossRef]
- Berry, V.; Pontikos, N.; Dudakova, L.; Moore, A.T.; Quinlan, R.; Liskova, P.; Michaelides, M. A novel missense mutation in LIM2 causing isolated autosomal dominant congenital cataract. Ophthalmic. Genet. 2020, 41, 131–134. [Google Scholar] [CrossRef]
- Merath, K.; Ronchetti, A.; Sidjanin, D.J. Functional analysis of HSF4 mutations found in patients with autosomal recessive congenital cataracts. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6646–6654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astiazarán, M.C.; García-Montaño, L.A.; Sánchez-Moreno, F.; Matiz-Moreno, H.; Zenteno, J.C. Next generation sequencing-based molecular diagnosis in familial congenital cataract expands the mutational spectrum in known congenital cataract genes. Am. J. Med. Genet. Part A 2018, 176, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Wang, J.D.; Jia, H.Y.; Zhang, J.S.; Li, Y.; Xiong, Y.; Li, J.; Li, X.X.; Huang, Y.; Zhu, G.Y.; et al. Mutation profiles of congenital cataract genes in 21 northern Chinese families. Mol. Vis. 2018, 24, 471–477. [Google Scholar]
- Ma, A.S.; Grigg, J.R.; Ho, G.; Prokudin, I.; Farnsworth, E.; Holman, K.; Cheng, A.; Billson, F.A.; Martin, F.; Fraser, C.; et al. Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing. Hum. Mutat. 2016, 37, 371–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Cao, Z.; Zhu, Y.; Liu, L.; Tong, Y.; Chen, X.; Wang, Y.; Lu, C.; Ma, X.; Yang, J. Mutation screening of crystallin genes in Chinese families with congenital cataracts. Mol. Vis. 2019, 25, 427–437. [Google Scholar] [PubMed]
- Micheal, S.; Niewold, I.T.G.; Siddiqui, S.N.; Zafar, S.N.; Khan, M.I.; Bergen, A.A.B. Delineation of novel autosomal recessive mutation in GJA3 and autosomal dominant mutations in GJA8 in Pakistani congenital cataract families. Genes 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome sequencing of 18 Chinese families with congenital cataracts: A new sight of the NHS gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef]
- Hansen, L.; Mikkelsen, A.; Nürnberg, P.; Nürnberg, G.; Anjum, I.; Eiberg, H.; Rosenberg, T. Comprehensive mutational screening in a cohort of danish families with hereditary congenital cataract. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3291–3303. [Google Scholar] [CrossRef]
- Anand, D.; Agrawal, S.A.; Slavotinek, A.; Lachke, S.A. Mutation Update of Transcription Factor Genes FOXE3, HSF4, MAF and PITX3 Causing Cataracts and Other Developmental Ocular Defects. Hum. Mutat. 2018, 39, 471–494. [Google Scholar] [CrossRef]
- Berry, V.; Pontikos, N.; Moore, A.; Ionides, A.C.W.; Plagnol, V.; Cheetham, M.E.; Michaelides, M. A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family. Eye 2018, 32, 806–812. [Google Scholar] [CrossRef]
- Jiao, X.; Khan, S.Y.; Kaul, H.; Butt, T.; Naeem, M.A.; Riazuddin, S.; Hejtmancik, J.F.; Riazuddin, S.A. Autosomal recessive congenital cataracts linked to HSF4 in a consanguineous Pakistani family. PLoS ONE 2019, 14, e0225010. [Google Scholar] [CrossRef]
- Dave, A.; Laurie, K.; Staffieri, S.E.; Taranath, D.; Mackey, D.A.; Mitchell, P.; Wang, J.J.; Craig, J.E.; Burdon, K.P.; Sharma, S. Mutations in the EPHA2 Gene Are a Major Contributor to Inherited Cataracts in South-Eastern Australia. PLoS ONE 2013, 8, e72518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, V.; Pontikos, N.; Albarca-Aguilera, M.; Plagnol, V.; Massouras, A.; Prescott, D.; Moore, A.T.; Arno, G.; Cheetham, M.E.; Michaelides, M. A recurrent splice-site mutation in EPHA2 causing congenital posterior nuclear cataract. Ophthalmic. Genet. 2018, 39, 236–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldahmesh, M.A.; Khan, A.O.; Mohamed, J.; Alkuraya, F.S. Novel recessive BFSP2 and PITX3 mutations: Insights into mutational mechanisms from consanguineous populations. Genet. Med. 2011, 13, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Addison, P.K.F.; Berry, V.; Ionides, A.C.W.; Francis, P.J.; Bhattacharya, S.S.; Moore, A.T. Posterior polar cataract is the predominant consequence of a recurrent mutation in the PITX3 gene. Br. J. Ophthalmol. 2005, 89, 138–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Li, D.; Cai, L.; Qiu, X.; Zhao, Z.; Wu, J.; Yang, J.; Lu, Y. A novel mutation in the OAR domain of PITX3 associated with congenital posterior subcapsular cataract. BMC Med. Genet. 2019, 20, 1–6. [Google Scholar] [CrossRef]
- Verdin, H.; Sorokina, E.A.; Meire, F.; Casteels, I.; De Ravel, T.; Semina, E.V.; De Baere, E. Novel and recurrent PITX3 mutations in Belgian families with autosomal dominant congenital cataract and anterior segment dysgenesis have similar phenotypic and functional characteristics. Orphanet. J. Rare Dis. 2014, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Type of CC | Microphthalmia | Microcornea | Iris Malformations | Family History of CC | Gene |
|---|---|---|---|---|---|---|
| Family 01 | Lamellar | - | - | - | - | CRYBB2 |
| Family 02 | Nuclear | Yes | - | - | - | CRYBA4 |
| Family 03 | Lamellar | - | - | - | Yes | CRYGS |
| Family 04 | Nuclear | Yes | - | - | Yes | CRYAA |
| Family 05 | Nuclear | - | - | - | Yes | CRYGD |
| Family 06 | Unknown | - | - | - | - | CRYGD |
| Family 07 | Nuclear | Yes | - | Yes | - | CRYGC |
| Family 08 | Nuclear | - | - | Yes | Yes | CRYGC |
| Family 09 | Lamellar | - | - | - | Yes | CRYBB3 |
| Family 10 | Lamellar | - | - | - | Yes | GJA3 |
| Family 11 | Nuclear | - | - | - | Yes | GJA3 |
| Family 12 | Nuclear | - | Yes | - | - | GJA8 |
| Family 13 | Lamellar | - | - | - | Yes | GJA8 |
| Family 14 | Nuclear | - | - | - | - | GJA8 |
| Family 15 | Posterior subcapsular | - | Yes | - | - | GJA8 |
| Family 16 | Nuclear | Yes | - | - | - | GJA8 |
| Family 17 | Nuclear | - | - | - | Yes | LIM2 |
| Family 18 | Lamellar | - | - | - | Yes | LIM2 |
| Family 19 | Unknown | - | - | - | - | LIM2 |
| Family 20 | Nuclear | Yes | - | - | Yes | EPHA2 |
| Family 21 | Unknown | - | - | - | - | EPHA2 |
| Family 22 | Nuclear | - | - | - | - | PAX6 |
| Family 23 | Unknown | Yes | Yes | - | - | PAX6 |
| Family 24 | Nuclear | - | - | - | Yes | MIP |
| Family 25 | Nuclear | - | - | - | Yes | MIP |
| Family 26 | Nuclear | - | - | - | Yes | MIP |
| Family 27 | Nuclear | - | - | - | Yes | HSF4 |
| Family 28 | Posterior subcapsular | - | - | - | Yes | PITX3 |
| Family 29 | Lamellar | - | - | - | - | ABCB6 |
| Family 30 | Posterior polar | - | - | - | - | TDRD7 |
| Family ID | Gene | Transcript | Mutation | ACMG Criteria | Variant Type | Zygosity | Segregation Analysis Performed | De Novo/Inherited | Described by |
|---|---|---|---|---|---|---|---|---|---|
| Family 01 | CRYBB2 | NM_000496.2 | c.562C>A:p.Arg188Ser | LP | Missense | Het | Yes | De novo | Wang Z et al., 2020 |
| Family 02 | CRYBA4 | NM_001886.3 | c.206T>C:p.Leu69Pro | P | Missense | Het | Yes | De novo | Billingsley G et al., 2006 |
| Family 03 | CRYGS | NM_017541 | c.53G>A:p.Gly18Asp | P | Missense | Het | Yes | Maternal | Zhai Y et al., 2017 |
| Family 04 | CRYAA | NM_000394.4 | c.61C>T:p.Arg21Trp | P | Missense | Het | Yes | Paternal | Hansen L et al., 2007 |
| Family 05 | CRYGD | NM_006891.3 | c.T232C:p.Ser78Pro | LP | Missense | Het | Yes | Maternal | Yang G et al., 2016 |
| Family 06 | CRYGD | NM_006891.3 | c.70C>T:p.Pro24Ser | P | Missense | Het | No | Unknown | Plotnikova OV et al., 2007 |
| Family 07 | CRYGC | NM_020989.4 | c.425_432dup:p.Leu145Glyfs * 5 | LP | Frameshift | Het | Yes | De novo | Graw J et al., 2002 |
| Family 08 | CRYGC | NM_020989.4 | c.438delG:p.Arg147Glyfs * 32 | LP | Frameshift | Het | Yes | Maternal | Novel |
| Family 09 | CRYBB3 | NM_004076.5 | c.531G>T:p.Glu177Asp | VUS | Missense | Het | Yes | Paternal | VCV000900831.1. Variation ID:900831 |
| Family 10 | GJA3 | NM_021954.4 | c.595G>A:p.Glu199Lys | LP | Missense | Het | Yes | Maternal | Novel |
| Family 11 | GJA3 | NM_021954.4 | c.817_818insATG:p.Tyr272_Ala273insAsp | LP | In-frame deletion | Het | Yes | Paternal | Novel |
| Family 12 | GJA8 | NM_005267.5 | c.226C>G:p.Arg76Gly | LP | Missense | Het | Yes | De novo | Reis LM et al., 2013 |
| Family 13 | GJA8 | NM_005267.5 | c.64G>A:p.Gly22Ser | LP | Missense | Het | Yes | Maternal | Ye Y et al., 2019 |
| Family 14 | GJA8 | NM_005267.5 | c.565C>G:p.Pro189Ala | LP | Missense | Het | No | Unknown | Novel |
| Family 15 | GJA8 | NM_005267.5 | c.226C>T:p.Arg76Cys | LP | Missense | Het | Yes | De novo | Reis LM et al., 2013 |
| Family 16 | GJA8 | NM_005267.5 | c.592C>T:p.Arg198Trp | P | Missense | Het | Yes | De novo | Hu S et al., 2010 |
| Family 17 | LIM2 | NM_030657.4 | c.388C>T:p.Arg130Cys | LP | Missense | Het | Yes | Paternal | Berry V et al., 2020 |
| Family 18 | LIM2 | NM_030657.4 | c.388C>T:p.Arg130Cys | LP | Missense | Het | Yes | Paternal | Berry V et al., 2020 |
| Family 19 | LIM2 | NM_030657.4 | c.385C>T:p.Arg129Cys | VUS | Missense | Het | Yes | Maternal | Novel |
| Family 20 | EPHA2 | NM_004431.4 | c.2826-9G>A | LP | Splice | Het | Yes | Maternal | Zhang T et al., 2009 |
| Family 21 | EPHA2 | NM_004431.4 | c.649G>C:p.Gly217Arg | VUS | Missense | Het | Yes | Paternal | Novel |
| Family 22 | PAX6 | NM_001258462.3 | c.77G>A:p.Arg26Gln | P | Missense | Het | Yes | De novo | Williamson KA et al., 2020 |
| Family 23 | PAX6 | NM_001258462.3 | c.219G>T:p.Arg73Ser | LP | Missense | Het | No | Unknown | Novel |
| Family 24 | MIP | NM_012064.3 | c.676dupC:p.Arg226fs | P | Frameshift | Het | Yes | Paternal | Novel |
| Family 25 | MIP | NM_012064.3 | c.430T>C:p.Cys144Arg | LP | Missense | Het | Yes | Maternal | Sun W et al., 2020 |
| Family 26 | MIP | NM_012064.3 | c.607-1G>T | LP | Splice | Het | Yes | De novo | Sun W et al., 2020 |
| Family 27 | HSF4 | NM_001040667.2 | Allele 1: c.486-2A>G Allele 2: c.1302delG:p.Leu436 | LP | Allele 1: Splice Allele 2: Frameshift | Compound het | Yes | Allele 1: Maternal Allele 2: Paternal | Novel |
| Family 28 | PITX3 | NM_005029.3 | c.640_656delGCCCTGCAGGGCCTGGG:p.Ala214Argfs * 42 | LP | Frameshift | Het | Yes | Paternal | Anand D et al., 2018 |
| Family 29 | ABCB6 | NM_005689.4 | c.1762G>A:p.Gly588Ser | VUS | Missense | Het | Yes | Maternal | Saison C et al., 2013 |
| Family 30 | TDRD7 | NM_014290.2 | Allele 1: c.1085C>T:p.Pro362Leu Allele 2: Not found | VUS | Allele 1: Missense Allele 2: Not found | Unknown | Yes | Allele 1: Maternal Allele 2: Not found | Novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Alcalde, C.; Nieves-Moreno, M.; Noval, S.; Peralta, J.M.; Montaño, V.E.F.; del Pozo, Á.; Santos-Simarro, F.; Vallespín, E. Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families. Genes 2021, 12, 580. https://doi.org/10.3390/genes12040580
Fernández-Alcalde C, Nieves-Moreno M, Noval S, Peralta JM, Montaño VEF, del Pozo Á, Santos-Simarro F, Vallespín E. Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families. Genes. 2021; 12(4):580. https://doi.org/10.3390/genes12040580
Chicago/Turabian StyleFernández-Alcalde, Celia, María Nieves-Moreno, Susana Noval, Jesús M. Peralta, Victoria E. F. Montaño, Ángela del Pozo, Fernando Santos-Simarro, and Elena Vallespín. 2021. "Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families" Genes 12, no. 4: 580. https://doi.org/10.3390/genes12040580
APA StyleFernández-Alcalde, C., Nieves-Moreno, M., Noval, S., Peralta, J. M., Montaño, V. E. F., del Pozo, Á., Santos-Simarro, F., & Vallespín, E. (2021). Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families. Genes, 12(4), 580. https://doi.org/10.3390/genes12040580