
Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome

 , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethics
2.2. Genomic DNA Extraction
2.3. DNA Methylation Analysis
2.4. SNP Array
2.5. Microsatellite Analysis
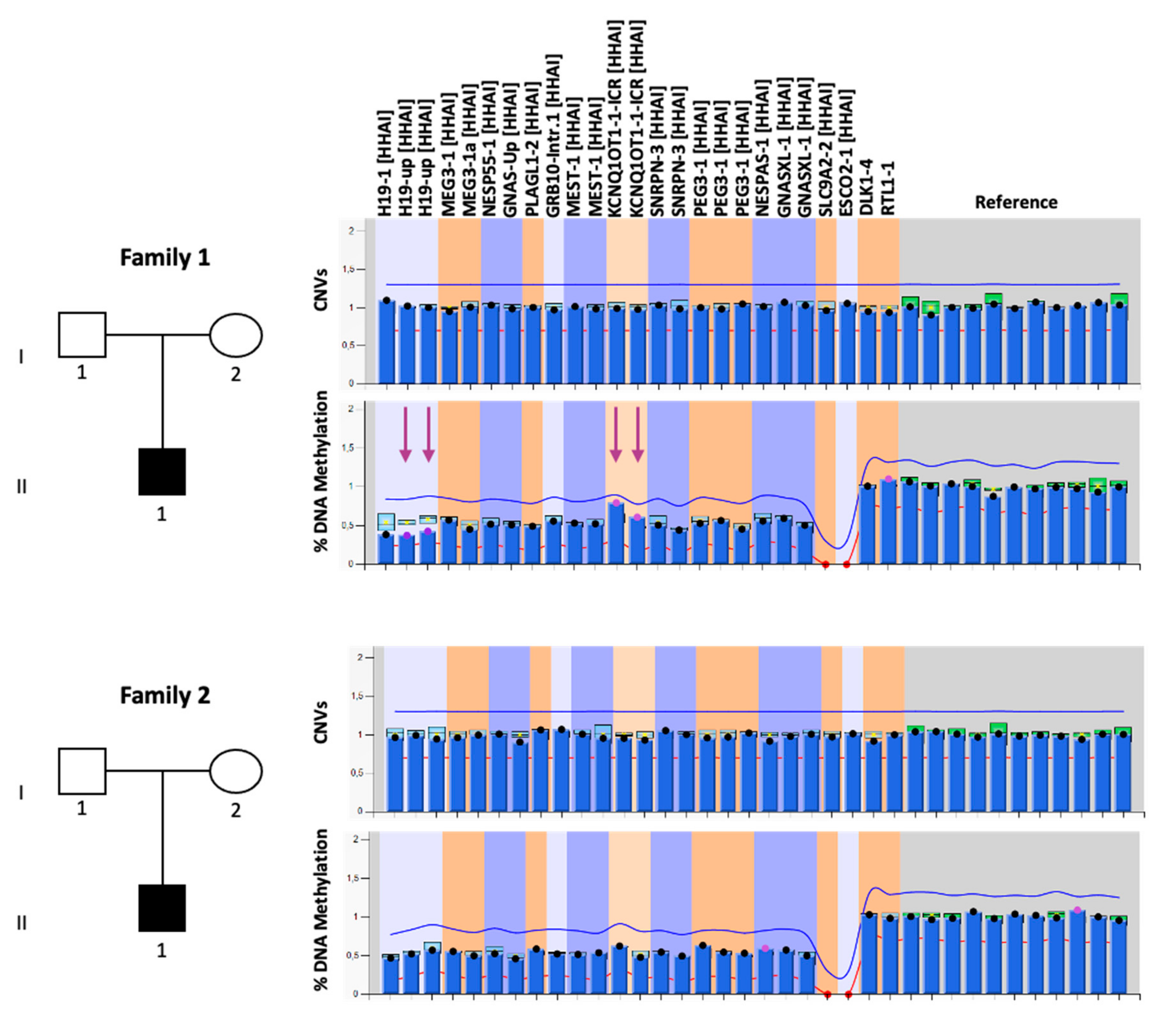
3. Results
3.1. Clinical Cases
3.2. Molecular Diagnosis
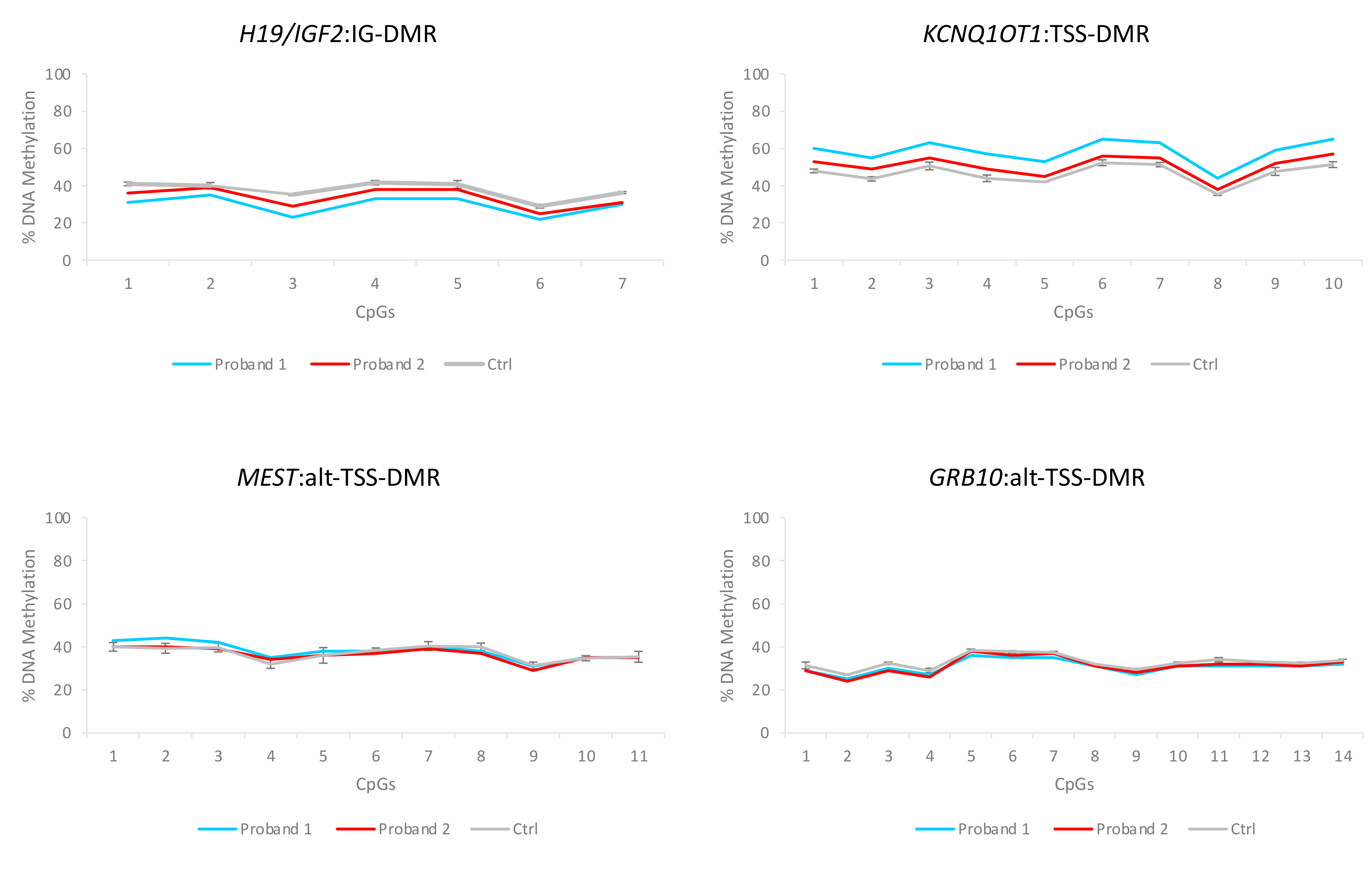
3.3. Characterization of the Molecular Defects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Azzi, S.; Salem, J.; Thibaud, N.; Chantot-Bastaraud, S.; Lieber, E.; Netchine, I.; Harbison, M.D. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J. Med. Genet. 2015, 52, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Soellner, L.; Kraft, F.; Sauer, S.; Begemann, M.; Kurth, I.; Elbracht, M.; Eggermann, T. Search for cis-acting factors and maternal effect variants in Silver-Russell patients with ICR1 hypomethylation and their mothers. Eur. J. Hum. Genet. 2019, 27, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzi, S.; Rossignol, S.; Steunou, V.; Sas, T.; Thibaud, N.; Danton, F.; Le Jule, M.; Heinrichs, C.; Cabrol, S.; Gicquel, C.; et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 2009, 18, 4724–4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brioude, F.; Oliver-Petit, I.; Blaise, A.; Praz, F.; Rossignol, S.; Le Jule, M.; Thibaud, N.; Faussat, A.M.; Tauber, M.; Le Bouc, Y.; et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 2013, 50, 823–830. [Google Scholar] [CrossRef]
- Mio, C.; Allegri, L.; Passon, N.; Bregant, E.; Demori, E.; Franzoni, A.; Driul, D.; Riccio, A.; Damante, G.; Baldan, F. A paternally inherited 1.4 kb deletion of the 11p15.5 imprinting center 2 is associated with a mild familial Silver-Russell syndrome phenotype. Eur. J. Hum. Genet. 2021, 29, 447–454. [Google Scholar] [CrossRef]
- Begemann, M.; Zirn, B.; Santen, G.; Wirthgen, E.; Soellner, L.; Büttel, H.M.; Schweizer, R.; van Workum, W.; Binder, G.; Eggermann, T. Paternally Inherited IGF2 Mutation and Growth Restriction. N. Engl. J. Med. 2015, 373, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Bullman, H.; Lever, M.; Robinson, D.O.; Mackay, D.J.; Holder, S.E.; Wakeling, E.L. Mosaic maternal uniparental disomy of chromosome 11 in a patient with Silver-Russell syndrome. J. Med. Genet. 2008, 45, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Luk, H.M.; Ivan Lo, F.M.; Sano, S.; Matsubara, K.; Nakamura, A.; Ogata, T.; Kagami, M. Silver-Russell syndrome in a patient with somatic mosaicism for upd(11)mat identified by buccal cell analysis. Am. J. Med. Genet. A 2016, 170, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- De Crescenzo, A.; Citro, V.; Freschi, A.; Sparago, A.; Palumbo, O.; Cubellis, M.V.; Carella, M.; Castelluccio, P.; Cavaliere, M.L.; Cerrato, F.; et al. A splicing mutation of the HMGA2 gene is associated with Silver-Russell syndrome phenotype. J. Hum. Genet. 2015, 60, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi Habib, W.; Brioude, F.; Edouard, T.; Bennett, J.T.; Lienhardt-Roussie, A.; Tixier, F.; Salem, J.; Yuen, T.; Azzi, S.; Le Bouc, Y.; et al. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet. Med. 2018, 20, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Poole, R.L.; Docherty, L.E.; Al Sayegh, A.; Caliebe, A.; Turner, C.; Baple, E.; Wakeling, E.; Harrison, L.; Lehmann, A.; Temple, I.K.; et al. Targeted methylation testing of a patient cohort broadens the epigenetic and clinical description of imprinting disorders. Am. J. Med. Genet. A 2013, 161A, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Kagami, M.; Mizuno, S.; Matsubara, K.; Nakabayashi, K.; Sano, S.; Fuke, T.; Fukami, M.; Ogata, T. Epimutations of the IG-DMR and the MEG3-DMR at the 14q32.2 imprinted region in two patients with Silver-Russell Syndrome-compatible phenotype. Eur. J. Hum. Genet. 2015, 23, 1062–1067. [Google Scholar] [CrossRef] [Green Version]
- Hjortshøj, T.D.; Sørensen, A.R.; Yusibova, M.; Hansen, B.M.; Dunø, M.; Balslev-Harder, M.; Grønskov, K.; van Hagen, J.M.; Polstra, A.M.; Eggermann, T.; et al. upd(20)mat is a rare cause of the Silver-Russell-syndrome-like phenotype: Two unrelated cases and screening of large cohorts. Clin. Genet. 2020, 97, 902–907. [Google Scholar] [CrossRef]
- Inoue, T.; Yagasaki, H.; Nishioka, J.; Nakamura, A.; Matsubara, K.; Narumi, S.; Nakabayashi, K.; Yamazawa, K.; Fuke, T.; Oka, A.; et al. Molecular and clinical analyses of two patients with UPD(16)mat detected by screening 94 patients with Silver-Russell syndrome phenotype of unknown aetiology. J. Med. Genet. 2019, 56, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Nakamura, A.; Iwahashi-Odano, M.; Tanase-Nakao, K.; Matsubara, K.; Nishioka, J.; Maruo, Y.; Hasegawa, Y.; Suzumura, H.; Sato, S.; et al. Contribution of gene mutations to Silver-Russell syndrome phenotype: Multigene sequencing analysis in 92 etiology-unknown patients. Clin. Epigenetics 2020, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Bliek, J.; Verde, G.; Callaway, J.; Maas, S.M.; De Crescenzo, A.; Sparago, A.; Cerrato, F.; Russo, S.; Ferraiuolo, S.; Rinaldi, M.M.; et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2009, 17, 611–619. [Google Scholar] [CrossRef]
- Bourque, D.K.; Avila, L.; Peñaherrera, M.; von Dadelszen, P.; Robinson, W.P. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010, 31, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Cubellis, M.V.; Pignata, L.; Verma, A.; Sparago, A.; Del Prete, R.; Monticelli, M.; Calzari, L.; Antona, V.; Melis, D.; Tenconi, R.; et al. Loss-of-function maternal-effect mutations of PADI6 are associated with familial and sporadic Beckwith-Wiedemann syndrome with multi-locus imprinting disturbance. Clin. Epigenetics 2020, 12, 139. [Google Scholar] [CrossRef]
- Palumbo, O.; Fichera, M.; Palumbo, P.; Rizzo, R.; Mazzolla, E.; Cocuzza, D.M.; Carella, M.; Mattina, T. TBR1 is the candidate gene for intellectual disability in patients with a 2q24.2 interstitial deletion. Am. J. Med. Genet. A 2014, 164A, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Papenhausen, P.; Schwartz, S.; Risheg, H.; Keitges, E.; Gadi, I.; Burnside, R.D.; Jaswaney, V.; Pappas, J.; Pasion, R.; Friedman, K.; et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am. J. Med. Genet. A 2011, 155A, 757–768. [Google Scholar] [CrossRef]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Cooper, W.N.; Curley, R.; Macdonald, F.; Maher, E.R. Mitotic recombination and uniparental disomy in Beckwith-Wiedemann syndrome. Genomics 2007, 89, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuka, Y.; Higashimoto, K.; Oka, T.; Yatsuki, H.; Jozaki, K.; Maeda, T.; Kawahara, K.; Hamasaki, Y.; Matsuo, M.; Nishioka, K.; et al. Identification of consensus motifs associated with mitotic recombination and clinical characteristics in patients with paternal uniparental isodisomy of chromosome 11. Hum. Mol. Genet. 2016, 25, 1406–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggermann, T. Prenatal Detection of Uniparental Disomies (UPD): Intended and Incidental Finding in the Era of Next Generation Genomics. Genes (Basel) 2020, 11, 1454. [Google Scholar] [CrossRef] [PubMed]
- Kotzot, D. Complex and segmental uniparental disomy (UPD): Review and lessons from rare chromosomal complements. J. Med. Genet. 2001, 38, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, F.Z.; Feldman, G.L.; McCaskill, C.; Subramanian, S.; Hughes, M.R.; Shaffer, L.G. Single cell analysis demonstrating somatic mosaicism involving 11p in a patient with paternal isodisomy and Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 1995, 4, 395–399. [Google Scholar] [CrossRef]
- Bonaglia, M.C.; Giorda, R.; Beri, S.; Bigoni, S.; Sensi, A.; Baroncini, A.; Capucci, A.; De Agostini, C.; Gwilliam, R.; Deloukas, P.; et al. Mosaic 22q13 deletions: Evidence for concurrent mosaic segmental isodisomy and gene conversion. Eur. J. Hum. Genet. 2009, 17, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Reik, W.; Brown, K.W.; Slatter, R.E.; Sartori, P.; Elliott, M.; Maher, E.R. Allelic methylation of H19 and IGF2 in the Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 1994, 3, 1297–1301. [Google Scholar] [CrossRef]
- Dutly, F.; Baumer, A.; Kayserili, H.; Yüksel-Apak, M.; Zerova, T.; Hebisch, G.; Schinzel, A. Seven cases of Wiedmann-Beckwith syndrome, including the first reported case of mosaic paternal isodisomy along the whole chromosome 11. Am. J. Med. Genet. 1998, 79, 347–353. [Google Scholar] [CrossRef]
- Romanelli, V.; Meneses, H.N.; Fernández, L.; Martínez-Glez, V.; Gracia-Bouthelier, R.; F Fraga, M.; Guillén, E.; Nevado, J.; Gean, E.; Martorell, L.; et al. Beckwith-Wiedemann syndrome and uniparental disomy 11p: Fine mapping of the recombination breakpoints and evaluation of several techniques. Eur. J. Hum. Genet. 2011, 19, 416–421. [Google Scholar] [CrossRef]
- Heide, S.; Chantot-Bastaraud, S.; Keren, B.; Harbison, M.D.; Azzi, S.; Rossignol, S.; Michot, C.; Lackmy-Port Lys, M.; Demeer, B.; Heinrichs, C.; et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J. Med. Genet. 2018, 55, 205–213. [Google Scholar] [CrossRef]
- Chiesa, N.; De Crescenzo, A.; Mishra, K.; Perone, L.; Carella, M.; Palumbo, O.; Mussa, A.; Sparago, A.; Cerrato, F.; Russo, S.; et al. The KCNQ1OT1 imprinting control region and non-coding RNA: New properties derived from the study of Beckwith-Wiedemann syndrome and Silver-Russell syndrome cases. Hum. Mol. Genet. 2012, 21, 10–25. [Google Scholar] [CrossRef]
- Nakashima, S.; Kato, F.; Kosho, T.; Nagasaki, K.; Kikuchi, T.; Kagami, M.; Fukami, M.; Ogata, T. Silver-Russell syndrome without body asymmetry in three patients with duplications of maternally derived chromosome 11p15 involving CDKN1C. J. Hum. Genet. 2015, 60, 91–95. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| STR | Family 1 | Family 2 |
|---|---|---|
| D11S922 | bc*, ac, bd | aa, ab, ab |
| TH | a*c, ab, cd | b*c, ab, cc |
| D11S4088 | a*c, ab, cd | c*d, ac, bd |
| Clinical Sign | Proband 1 (Segmental) | Proband 2 (Whole-Chromosome) | Proband Described in [8] (Whole-Chromosome) | Proband Described in [9] (Whole-Chromosome) | 11p15 LoM (%) * | Upd(7)mat (%) * | 11p15 dup (%) |
|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Male | |||
| 1 SGA: birth weight and/or birth length | Yes | Yes | Yes | Yes | 100 | 73 | 100 ** |
| Postnatal growth failure | Yes | Yes | Yes | Yes | 84 | 81 | 100 ** |
| Relative macrocephaly at birth | Yes | Yes | No | Yes | 99 | 85 | 95 ** |
| Protruding forehead | Yes | Yes | Yes | Yes | 94 | 100 | 90 ** |
| Body asymmetry | Yes | Yes | Yes | Yes | 77 | 29 | 0 ** |
| Feeding difficulties and/or low BMI | Yes | Yes | Yes | Yes | 72 | 87 | 88 ** |
| Triangular face | Yes | Yes | Yes | Yes | 99 | 50 | 74 *** |
| Fifth finger clinodactyly | Yes | No | Yes | Yes | 81 | 56 | 93 *** |
| Micrognathia | Yes | No | No | No | 75 | 26 | nr *** |
| Low muscle mass | Yes | Yes | No | No | 67 | 47 | 67 *** |
| Excessive sweating | No | Yes | Yes | No | 51 | 70 | nr *** |
| Down-turned mouth | Yes | No | No | No | 57 | 26 | nr *** |
| Genital abnormalities | No | Yes | No | Yes | nr | nr | nr *** |
| Speech delay | Yes | No | No | Yes | 32 | 64 | 78 *** |
| Motor delay | Yes | Yes | Yes | No | 30 | 58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignata, L.; Sparago, A.; Palumbo, O.; Andreucci, E.; Lapi, E.; Tenconi, R.; Carella, M.; Riccio, A.; Cerrato, F. Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome. Genes 2021, 12, 581. https://doi.org/10.3390/genes12040581
Pignata L, Sparago A, Palumbo O, Andreucci E, Lapi E, Tenconi R, Carella M, Riccio A, Cerrato F. Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome. Genes. 2021; 12(4):581. https://doi.org/10.3390/genes12040581
Chicago/Turabian StylePignata, Laura, Angela Sparago, Orazio Palumbo, Elena Andreucci, Elisabetta Lapi, Romano Tenconi, Massimo Carella, Andrea Riccio, and Flavia Cerrato. 2021. "Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome" Genes 12, no. 4: 581. https://doi.org/10.3390/genes12040581
APA StylePignata, L., Sparago, A., Palumbo, O., Andreucci, E., Lapi, E., Tenconi, R., Carella, M., Riccio, A., & Cerrato, F. (2021). Mosaic Segmental and Whole-Chromosome Upd(11)mat in Silver-Russell Syndrome. Genes, 12(4), 581. https://doi.org/10.3390/genes12040581