Congenital Microcoria: Clinical Features and Molecular Genetics



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
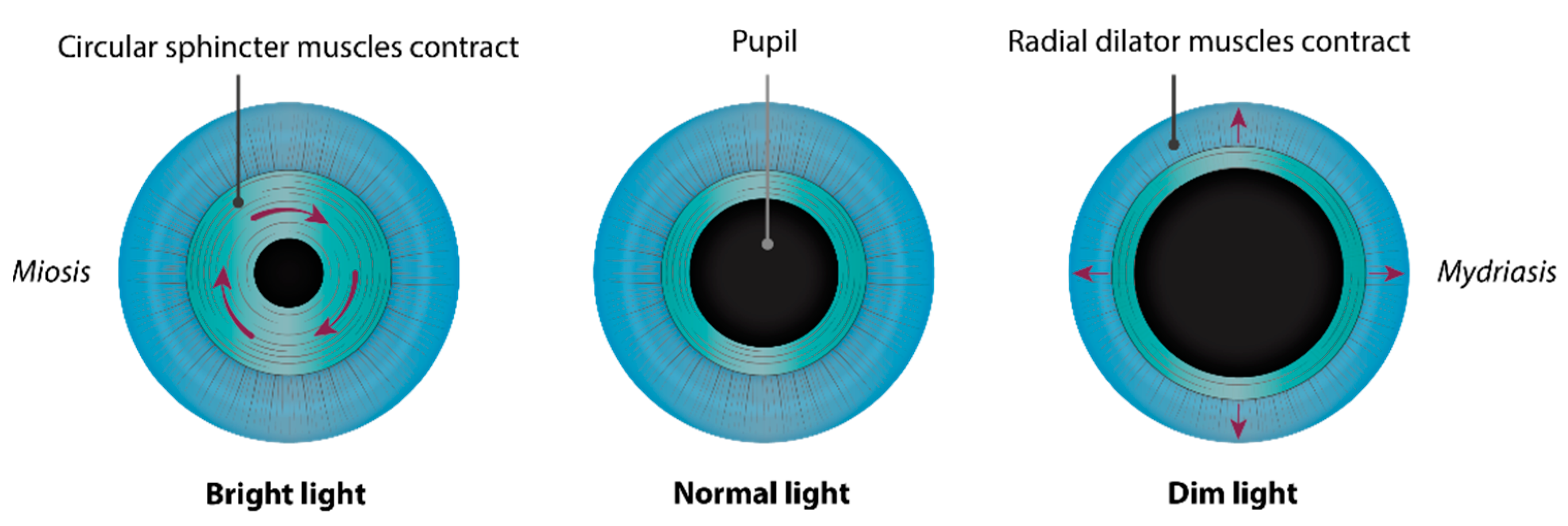
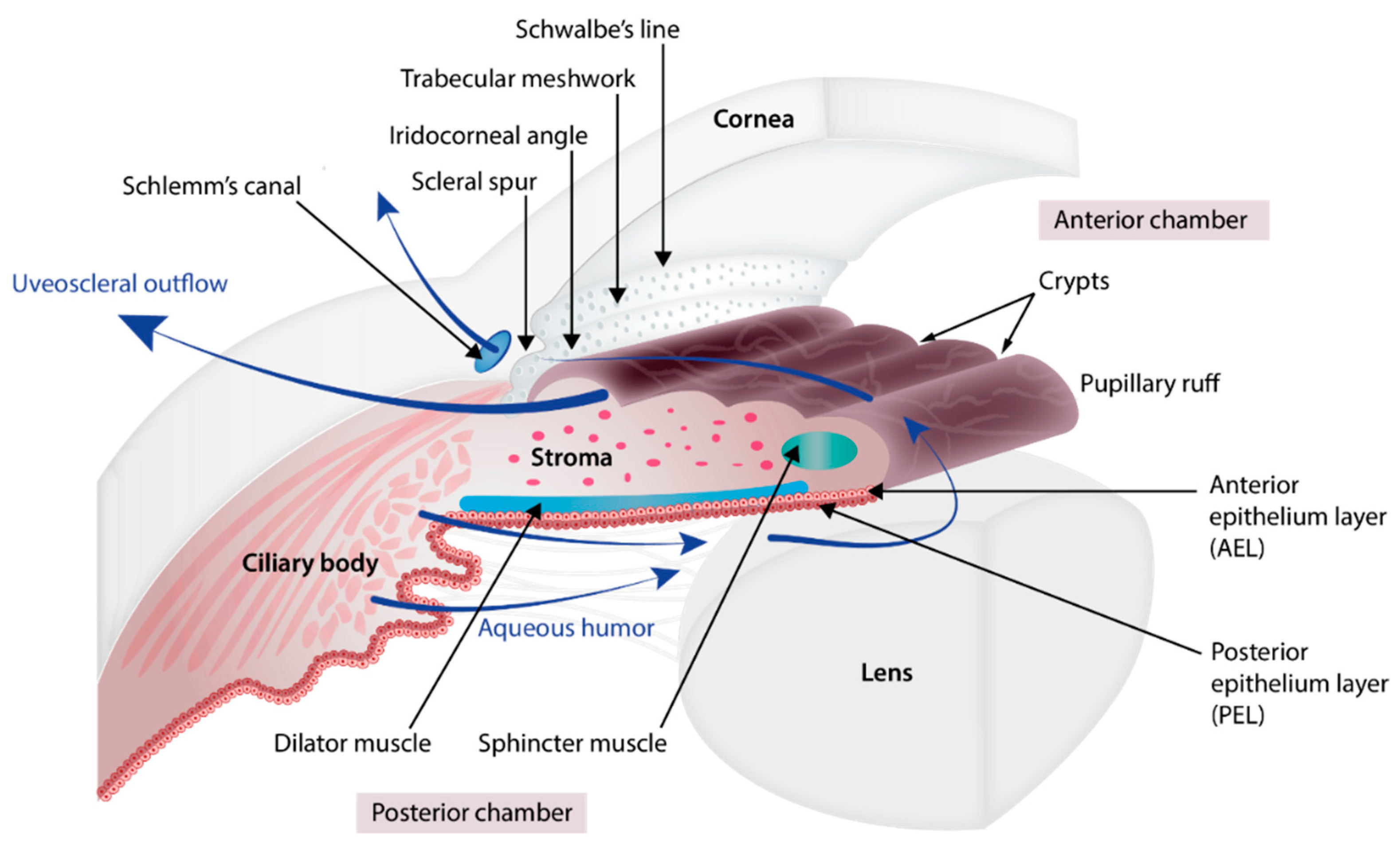
2. Embryology of the Chamber Angle and Iris
3. Disease Description
3.1. Associated Signs
3.1.1. Glaucoma
3.1.2. Axial Myopia
3.1.3. Astigmatism and Other Corneal Anomalies
3.1.4. Cataract
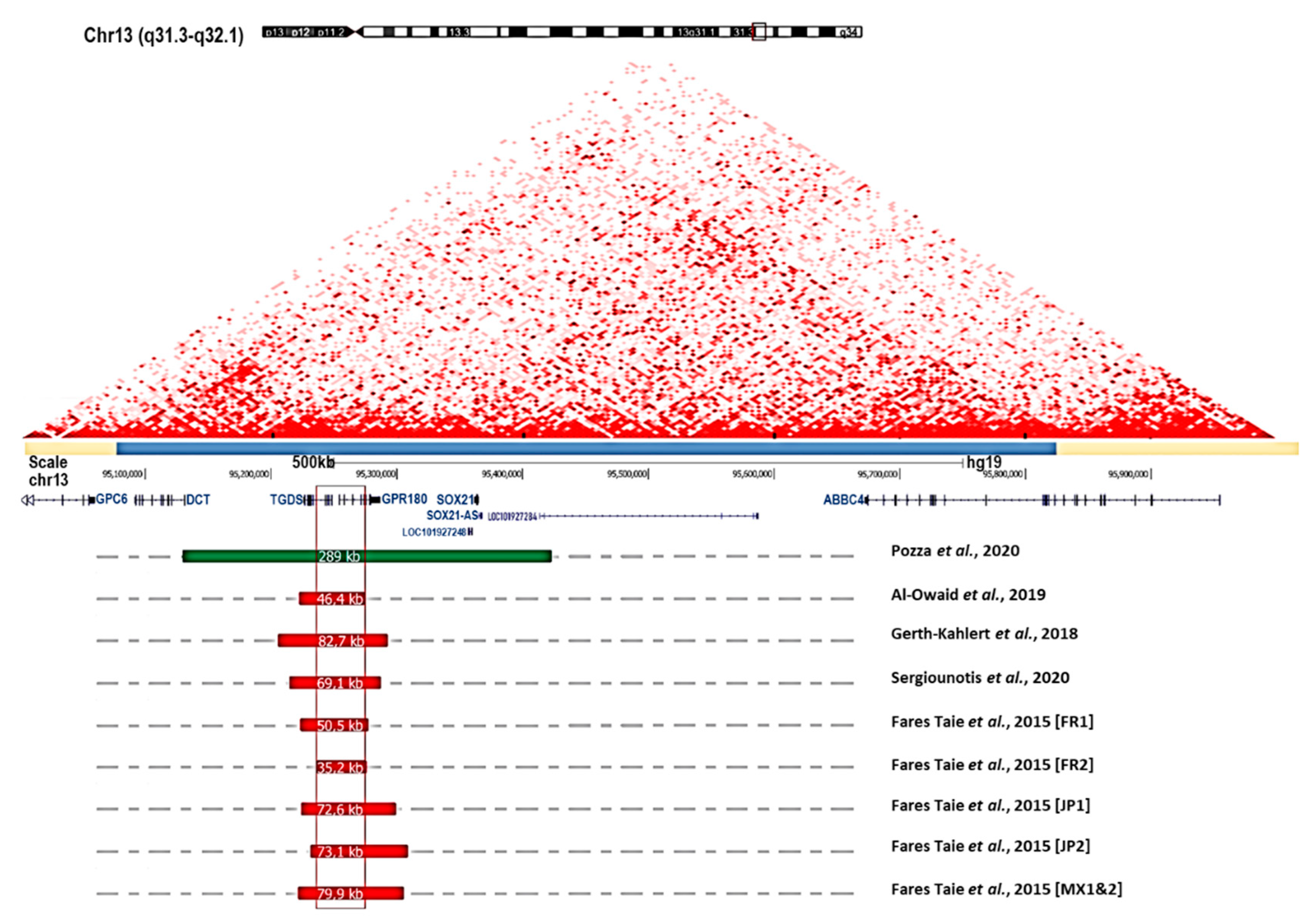
4. Genetics
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, N.; Ashery-Padan, R. Iris Development in Vertebrates; Genetic and Molecular Considerations. Brain Res. 2008, 1192, 17–28. [Google Scholar] [CrossRef]
- Bloom, J.; Motlagh, M.; Czyz, C.N. Anatomy, Head and Neck, Eye Iris Sphincter Muscle. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Uvea. Clinical Gate 2015. Available online: https://clinicalgate.com/the-uveal-tract/ (accessed on 8 March 2015).
- Forrester, J.; Dick, A.; McMenamin, P.; Roberts, F.; Pearlman, E. The Eye; Elsevier: Amsterdam, The Netherlands, 2016; ISBN1 978-0-7020-5554-6. Available online: https://www.sciencedirect.com/book/9780702055546/the-eye (accessed on 19 February 2015)ISBN2 978-0-7020-5554-6.
- Abu-Hassan, D.W.; Acott, T.S.; Kelley, M.J. The Trabecular Meshwork: A Basic Review of Form and Function. J. Ocul. Biol. 2014, 2, 1–22. [Google Scholar] [CrossRef]
- Goel, M.; Picciani, R.G.; Lee, R.K.; Bhattacharya, S.K. Aqueous Humor Dynamics: A Review. Open Ophthalmol. J. 2010, 4, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Fares-Taie, L.; Gerber, S.; Tawara, A.; Ramirez-Miranda, A.; Douet, J.-Y.; Verdin, H.; Guilloux, A.; Zenteno, J.C.; Kondo, H.; Moisset, H.; et al. Submicroscopic Deletions at 13q32.1 Cause Congenital Microcoria. Am. J. Hum. Genet. 2015, 96, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Toulemont, P.J.; Urvoy, M.; Coscas, G.; Lecallonnec, A.; Cuvilliers, A.F. Association of Congenital Microcoria with Myopia and Glaucoma. Ophthalmology 1995, 102, 193–198. [Google Scholar] [CrossRef]
- Tawara, A.; Itou, K.; Kubota, T.; Harada, Y.; Tou, N.; Hirose, N. Congenital Microcoria Associated With Late-Onset Developmental Glaucoma. J. Glaucoma 2005, 14, 409–413. [Google Scholar] [CrossRef]
- Rouillac, C.; Roche, O.; Marchant, D.; Bachner, L.; Kobetz, A.; Toulemont, P.-J.; Orssaud, C.; Urvoy, M.; Odent, S.; Le Marec, B.; et al. Mapping of a Congenital Microcoria Locus to 13q31-Q32. Am. J. Hum. Genet. 1998, 62, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, W.A.C. The Ultrastructural Pathological Features of Congenital Microcoria: A Case Report. Arch. Ophthalmol. 1989, 107, 99. [Google Scholar] [CrossRef] [PubMed]
- Pozza, E.; Verdin, H.; Deconinck, H.; Dheedene, A.; Menten, B.; De Baere, E.; Balikova, I. Microcoria Due to First Duplication of 13q32.1 Including the GPR180 Gene and Maternal Mosaicism. Eur. J. Med. Genet 2020, 63, 103918. [Google Scholar] [CrossRef] [PubMed]
- Pierson, M.; Cordier, J.; Hervouuet, F.; Rauber, G. An Unusual Congenital and Familial Congenital Malformative Combination Involving the Eye and KidneY. J. Genet Hum. 1963, 12, 184–213. [Google Scholar]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Müntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wühl, E.; et al. Human Laminin Β2 Deficiency Causes Congenital Nephrosis with Mesangial Sclerosis and Distinct Eye Abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef]
- Wilde, W.R. An Essay on the Malformations and Congenital Diseases of the Organs of Sight/by W. R. Wilde; John Churchill: London, UK, 1862. [Google Scholar]
- Edward, D.P.; Kaufman, L.M. Anatomy, Development, and Physiology of the Visual System. Pediatr. Clin. N. Am. 2003, 50, 1–23. [Google Scholar] [CrossRef]
- Cvekl, A.; Tamm, E.R. Anterior Eye Development and Ocular Mesenchyme. Bioessays 2004, 26, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Bales, T.R.; Lopez, M.J.; Clark, J. Embryology, Eye. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Graw, J. Eye Development. Curr. Top. Dev. Biol. 2010, 90, 343–386. [Google Scholar] [CrossRef]
- Ruprecht, K.W.; Wulle, K.G. Light and electron microscopic studies on the development of the human pupillary sphincter muscle. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. Albrecht Graefe’s Arch. Clin. Exp. Ophthalmol. 1973, 186, 117–130. [Google Scholar] [CrossRef]
- Polomeno, R.C.; Milot, J. Congenital Miosis. Can. J. Ophthalmol. 1979, 14, 43–46. [Google Scholar]
- Mann, I.C. Notes on the Anatomy of the Living Eye, as Revealed by the Gullstrand Slitlamp. J. Anat. 1925, 59, 155–165. [Google Scholar]
- Anderson, D.R. The Development of the Trabecular Meshwork and Its Abnormality in Primary Infantile Glaucoma. Trans. Am. Ophthalmol. Soc. 1981, 79, 458–485. [Google Scholar] [PubMed]
- Vranka, J.A.; Kelley, M.J.; Acott, T.S.; Keller, K.E. Extracellular Matrix in the Trabecular Meshwork: Intraocular Pressure Regulation and Dysregulation in Glaucoma. Exp. Eye Res. 2015, 133, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treacher Collins, E.; Mayou, M.S. Pathology and Bacteriology in Ophthalmic Practice; William Heinemann: London, UK, 1911; pp. 64, 82. [Google Scholar]
- Wood, C.A. Fully Illustrated; Lenicet to Muscles, Ocular (Classic Reprint). In The American Encyclopedia and Dictionary of Ophthalmology; Forgotten Books: London, UK, 1917; Volume 10, ISBN 1396359575 (ISBN13: 9781396359576). [Google Scholar]
- Truc and Valude. Nouveaux Éléments d’Ophtalmologie; Maloine: Paris, France, 1896; p. 475. [Google Scholar]
- Ramirez-Miranda, A.; Paulin-Huerta, J.M.; Chavez-Mondragón, E.; Islas-de la Vega, G.; Rodriguez-Reyes, A. Ultrabiomicroscopic-Histopathologic Correlations in Individuals with Autosomal Dominant Congenital Microcoria: Three-Generation Family Report. Case Rep. Ophthalmol. 2011, 2, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Saxena, R.C. Congenital Microcoria: A Study in Three Generations. Indian J. Ophthalmol. 1993, 41, 130. [Google Scholar]
- Ramprasad, V.L.; Sripriya, S.; Ronnie, G.; Nancarrow, D.; Saxena, S.; Hemamalini, A.; Kumar, D.; Vijaya, L.; Kumaramanickavel, G. Genetic Homogeneity for Inherited Congenital Microcoria Loci in an Asian Indian Pedigree. Mol. Vis. 2005, 11, 934–940. [Google Scholar]
- Sergouniotis, P.I.; Ellingford, J.M.; O’Sullivan, J.; Fenerty, C.H.; Black, G.C. Genome Sequencing Identifies a Large Deletion at 13q32.1 as the Cause of Microcoria and Childhood-Onset Glaucoma. Acta Ophthalmol. 2017, 95, e249–e250. [Google Scholar] [CrossRef] [PubMed]
- Gerth-Kahlert, C.; Maggi, J.; Töteberg-Harms, M.; Tiwari, A.; Budde, B.; Nürnberg, P.; Koller, S.; Berger, W. Absence of Goniodysgenesis in Patients with Chromosome 13Q Microdeletion-Related Microcoria. Ophthalmol. Glaucoma 2018, 1, 145–147. [Google Scholar] [CrossRef] [Green Version]
- Holth, S.; Berner, O. Congenital miosis or pinhole pupils owing to developmental faults of the dilatator muscle. Br. J. Ophthalmol. 1923, 7, 401–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.M.; Raviola, G.; Miller, C.D.; Friedmann, A.I. Fine Structural Defects in a Case of Congenital Microcoria. Graefes Arch. Clin. Exp. Ophthalmol. 1989, 227, 88–94. [Google Scholar] [CrossRef]
- Ferreira, B.F.D.A.; Schmidt, M.B.; Barbosa, L.J.; Oyamada, M.K.; Carricondo, P.C. Phacoemulsification and 1% Atropine Eye Drops for Treatment of Antimetropic Congenital Microcoria Associated with Cataracts. Arq. Bras. Oftalmol. 2019, 82. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.R.; Amaya, L.; Taylor, D. Congenital Idiopathic Microcoria. Am. J. Ophthalmol. 1988, 106, 590–594. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Corvino, C.; DeBlasi, A.; Calabrò, F. Congenital Microcoria: Case Report and Histological Study. J. Pediatr. Ophthalmol. Strabismus. 1998, 35, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Bremner, F.D.; Houlden, H.; Smith, S.E. Genotypic and Phenotypic Heterogeneity in Familial Microcoria. Br. J. Ophthalmol. 2004, 88, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Meire, F.M.; Delleman, J.W. Autosomal Dominant Congenital Miosis with Megalocornea. Ophthalmic Paediatr. Genet. 1992, 13, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hyams, S.W.; Neumann, E. Congenital Microcoria and Combined Mechanism Glaucoma. Am. J. Ophthalmol. 1969, 68, 326–327. [Google Scholar] [CrossRef]
- Imaizumi, M.; Kuwabara, T. Development of the Rat Iris. Investig. Ophthalmol. 1971, 10, 733–744. [Google Scholar]
- Jensen, A.M. Potential Roles for BMP and Pax Genes in the Development of Iris Smooth Muscle. Dev. Dyn. 2005, 232, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Thumann, G. Development and Cellular Functions of the Iris Pigment Epithelium. Surv. Ophthalmol. 2001, 45, 345–354. [Google Scholar] [CrossRef]
- Mann, I.C. The Development of the Human Iris. Br. J. Ophthalmol. 1925, 9, 495–512. [Google Scholar] [CrossRef] [Green Version]
- Uusitalo, M.; Kivelä, T. Development of Cytoskeleton in Neuroectodermally Derived Epithelial and Muscle Cells of the Human Eye. Investig. Opthalmol. Vis. Sci. 1995, 36, 2584–2591. [Google Scholar]
- Mazzeo, V.; Gaiba, G.; Rossi, A. Hereditary Cases of Congenital Microcoria and Goniodysgenesis. Ophthalmic Paediatr. Genet. 1986, 7, 121–125. [Google Scholar] [CrossRef]
- Veirs, E.R. Congenital Miosis: Associated with a Narrow Angle of the Anterior Chamber and Abnormally Placed Iris Tissue. Arch. Ophthalmol. 1961, 65, 59. [Google Scholar] [CrossRef]
- Heatley, J. Miosis Congenita Familiar. Soc. Mex. Oftalmol. 1948, 22, 141–148. [Google Scholar]
- Ardouin, M.; Urvoy, M.; Lefranc, J. Microcorie Congenitale. Bull. Mem Soc. Fr. Ophtalmol. 1964, 77, 356. [Google Scholar]
- Stabilization of Glaucoma Associated with Microcoria|Semantic Scholar. Available online: https://www.semanticscholar.org/paper/Stabilization-of-Glaucoma-Associated-with-Ngabou/6ade8acf89a87b9afe174a5c2f10d7702229fcc4 (accessed on 9 March 2021).
- Sahori, A.; Katsumori, O.K. Congenital Miosis Associated with Juvenile Glaucoma. Folia Ophthalmol. Jpn. 1987, 38, 853–857. [Google Scholar]
- Coulon, G.; Delbosc, B.; Jeffredo, Y.; Viennet, G.; Oppermann, A.; Royer, J. Congenital microcoria: A case report with histopathological study. J. Fr. Ophtalmol. 1986, 9, 35–39. [Google Scholar]
- Hoskins, H.D.; Shaffer, R.N.; Hetherington, J. Anatomical Classification of the Developmental Glaucomas. Arch. Ophthalmol. 1984, 102, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Agoston, I.; Gróf, P. La Miose Congénitale et l’albinisme. OPH 1968, 155, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Acott, T.S.; Kelley, M.J. Extracellular Matrix in the Trabecular Meshwork. Exp. Eye Res. 2008, 86, 543–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, P.; Hourihan, F.; Sandbach, J.; Wang, J.J. The Relationship between Glaucoma and Myopia: The Blue Mountains Eye Study. Ophthalmology 1999, 106, 2010–2015. [Google Scholar] [CrossRef]
- Quinn, G.E.; Berlin, J.A.; Young, T.L.; Ziylan, S.; Stone, R.A. Association of Intraocular Pressure and Myopia in Children. Ophthalmology 1995, 102, 180–185. [Google Scholar] [CrossRef]
- Chen, S.-J.; Lu, P.; Zhang, W.-F.; Lu, J.-H. High Myopia as a Risk Factor in Primary Open Angle Glaucoma. Int. J. Ophthalmol. 2012, 5, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Tielsch, J.M. Risk Factors for Open-Angle Glaucoma: The Barbados Eye Study. JAMA Ophthalmol. 1996, 114. [Google Scholar] [CrossRef]
- Schmid, K.L.; Hills, T.; Abbott, M.; Humphries, M.; Pyne, K.; Wildsoet, C.F. Relationship between Intraocular Pressure and Eye Growth in Chick. Ophthalmic Physiol. Opt. 2003, 23, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.S.; Stone, R.D.; Fromer, C.; Billson, F.A. Monocular Axial Myopia Associated with Neonatal Eyelid Closure in Human Infants. Am. J. Ophthalmol. 1981, 91, 197–200. [Google Scholar] [CrossRef]
- Rabin, J.; Sluyters, R.C.V.; Malach, R. Emmetropization: A Vision-Dependent Phenomenon. Investig. Opthalmol. Vis. Sci. 1981, 20, 561–564. [Google Scholar]
- Johnson, C.A.; Post, R.B.; Chalupa, L.M.; Lee, T.J. Monocular Deprivation in Humans: A Study of Identical Twins. Investig. Opthalmol. Vis. Sci. 1982, 23, 135–138. [Google Scholar]
- Raviola, E.; Wiesel, T.N. An Animal Model of Myopia. N. Engl. J. Med. 1985, 312, 1609–1615. [Google Scholar] [CrossRef]
- Pardue, M.T.; Faulkner, A.E.; Fernandes, A.; Yin, H.; Schaeffel, F.; Williams, R.W.; Pozdeyev, N.; Iuvone, P.M. High Susceptibility to Experimental Myopia in a Mouse Model with a Retinal ON Pathway Defect. Investig. Opthalmol. Vis. Sci. 2008, 49, 706–712. [Google Scholar] [CrossRef]
- Schaeffel, F.; Feldkaemper, M. Animal Models in Myopia Research. Clin. Exp. Opt. 2015, 98, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Al-Owaid, A.; Alarfaj, M.; Al-Qahtani, A.; Al-Arfaj, K. Congenital Microcoria in a Saudi Family. Ophthalmic Genet. 2019, 40, 578–580. [Google Scholar] [CrossRef]
- Vail, D.T., Jr. Adult Hereditary Anterior Megalophthalmus Sine Glaucoma: A Definite Disease Entity: With Special Reference To The Extraction Of Cataract. Arch. Ophthalmol. 1931, 6, 39–62. [Google Scholar] [CrossRef]
- Van Leeuwen, M.A. La Microcorie Congenitale Hereditaire. Bull Soc Belge Ophtalmol 1949, 91, 118–136. [Google Scholar]
- Castilla-Vallmanya, L.; Gürsoy, S.; Giray-Bozkaya, Ö.; Prat-Planas, A.; Bullich, G.; Matalonga, L.; Centeno-Pla, M.; Rabionet, R.; Grinberg, D.; Balcells, S.; et al. De Novo PORCN and ZIC2 Mutations in a Highly Consanguineous Family. Int. J. Mol. Sci. 2021, 22, 1549. [Google Scholar] [CrossRef] [PubMed]
- Perrault, I.; Hanein, S.; Gerber, S.; Barbet, F.; Dufier, J.-L.; Munnich, A.; Rozet, J.-M.; Kaplan, J. Evidence of Autosomal Dominant Leber Congenital Amaurosis (LCA) Underlain by a CRX Heterozygous Null Allele. J. Med. Genet. 2003, 40, e90. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Song, F.; Zhang, B.; Zhang, L.; Xu, J.; Kuang, D.; Li, D.; Choudhary, M.N.K.; Li, Y.; Hu, M.; et al. The 3D Genome Browser: A Web-Based Browser for Visualizing 3D Genome Organization and Long-Range Chromatin Interactions. Genome Biol. 2018, 19, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costin, G.-E.; Valencia, J.C.; Wakamatsu, K.; Ito, S.; Solano, F.; Milac, A.L.; Vieira, W.D.; Yamaguchi, Y.; Rouzaud, F.; Petrescu, A.-J.; et al. Mutations in Dopachrome Tautomerase (Dct) Affect Eumelanin/Pheomelanin Synthesis, but Do Not Affect Intracellular Trafficking of the Mutant Protein. Biochem. J. 2005, 391, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennamen, P.; Tingaud-Sequeira, A.; Gazova, I.; Keighren, M.; McKie, L.; Marlin, S.; Halem, S.G.; Kaplan, J.; Delevoye, C.; Lacombe, D.; et al. Dopachrome Tautomerase Variants in Patients with Oculocutaneous Albinism. bioRxiv 2020. [Google Scholar] [CrossRef]
- Uchikawa, M.; Kamachi, Y.; Kondoh, H. Two Distinct Subgroups of Group B Sox Genes for Transcriptional Activators and Repressors: Their Expression during Embryonic Organogenesis of the Chicken. Mech. Dev. 1999, 84, 103–120. [Google Scholar] [CrossRef]
- Pauls, S.; Smith, S.F.; Elgar, G. Lens Development Depends on a Pair of Highly Conserved Sox21 Regulatory Elements. Dev. Biol. 2012, 365–248, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, P.P.; Pecen, P.E.; Rao, P.V. MRP4-Mediated Regulation of Intracellular CAMP and CGMP Levels in Trabecular Meshwork Cells and Homeostasis of Intraocular Pressure. Investig. Opthalmol. Vis. Sci. 2013, 54, 1636. [Google Scholar] [CrossRef]
- Carreon, T.; van der Merwe, E.; Fellman, R.L.; Johnstone, M.; Bhattacharya, S.K. Aqueous Outflow—A Continuum from Trabecular Meshwork to Episcleral Veins. Prog. Retin. Eye Res. 2017, 57, 108–133. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angée, C.; Nedelec, B.; Erjavec, E.; Rozet, J.-M.; Fares Taie, L. Congenital Microcoria: Clinical Features and Molecular Genetics. Genes 2021, 12, 624. https://doi.org/10.3390/genes12050624
Angée C, Nedelec B, Erjavec E, Rozet J-M, Fares Taie L. Congenital Microcoria: Clinical Features and Molecular Genetics. Genes. 2021; 12(5):624. https://doi.org/10.3390/genes12050624
Chicago/Turabian StyleAngée, Clémentine, Brigitte Nedelec, Elisa Erjavec, Jean-Michel Rozet, and Lucas Fares Taie. 2021. "Congenital Microcoria: Clinical Features and Molecular Genetics" Genes 12, no. 5: 624. https://doi.org/10.3390/genes12050624
APA StyleAngée, C., Nedelec, B., Erjavec, E., Rozet, J. -M., & Fares Taie, L. (2021). Congenital Microcoria: Clinical Features and Molecular Genetics. Genes, 12(5), 624. https://doi.org/10.3390/genes12050624