
The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD


Abstract
:1. Introduction
2. The Characterization and Identification of the PPARs and LXRs Nuclear Receptors
2.1. Peroxisome Proliferator-Activated Receptors (PPARs)
2.2. Liver X Receptor
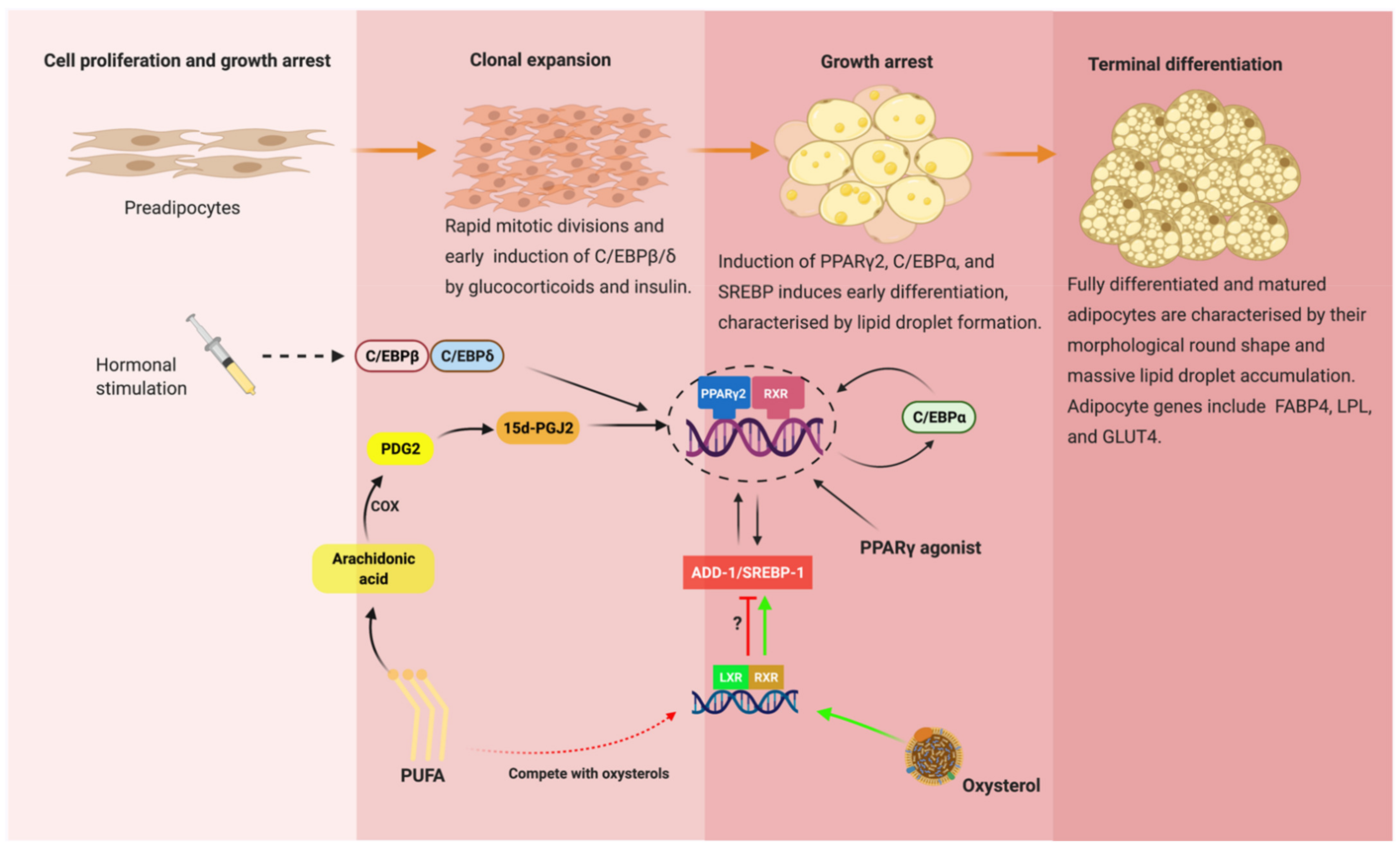
3. Transcriptional Regulation of Adipogenesis: Interplay between C/EBPs, PPARs and SREBPs
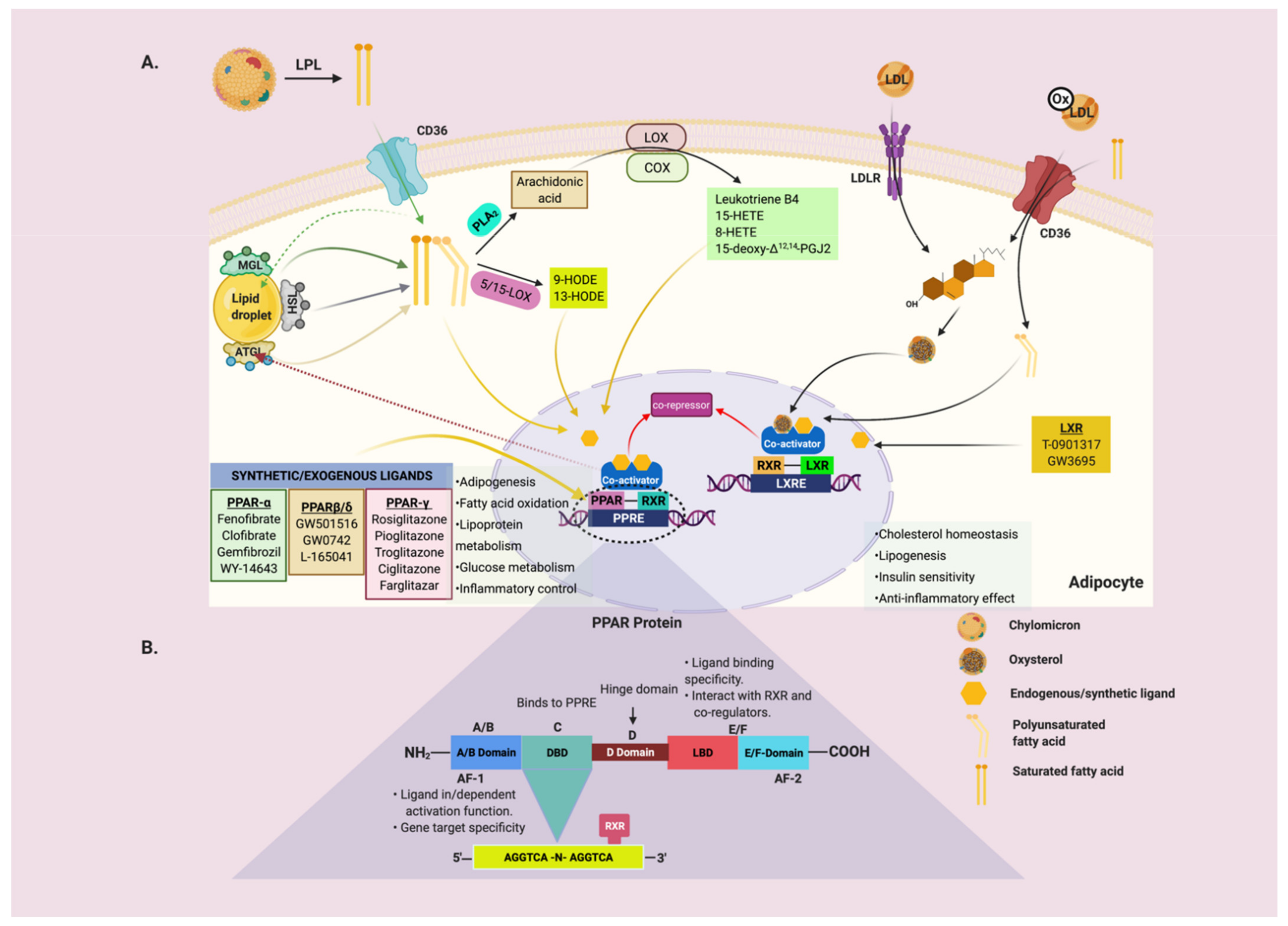
4. The Interdependent Role of Metabolic Lipases and Nuclear Receptors (PPARs and LXR)
4.1. Adipose Triglyceride Lipase (ATGL)
4.2. Hormone-Sensitive Lipase (HSL)
4.3. Monoglyceride Lipase (MGL)
4.4. Lipoprotein Lipase (LPL)
5. Physiological and Pharmacological Ligands for PPAR and LXR
5.1. Endogenous and Synthetic Ligands for PPARα
5.2. Endogenous and Synthetic Ligands for PPAR-β/δ
5.3. Endogenous and Synthetic Ligands for PPARγ
5.4. Endogenous and Synthetic Ligands for LXR
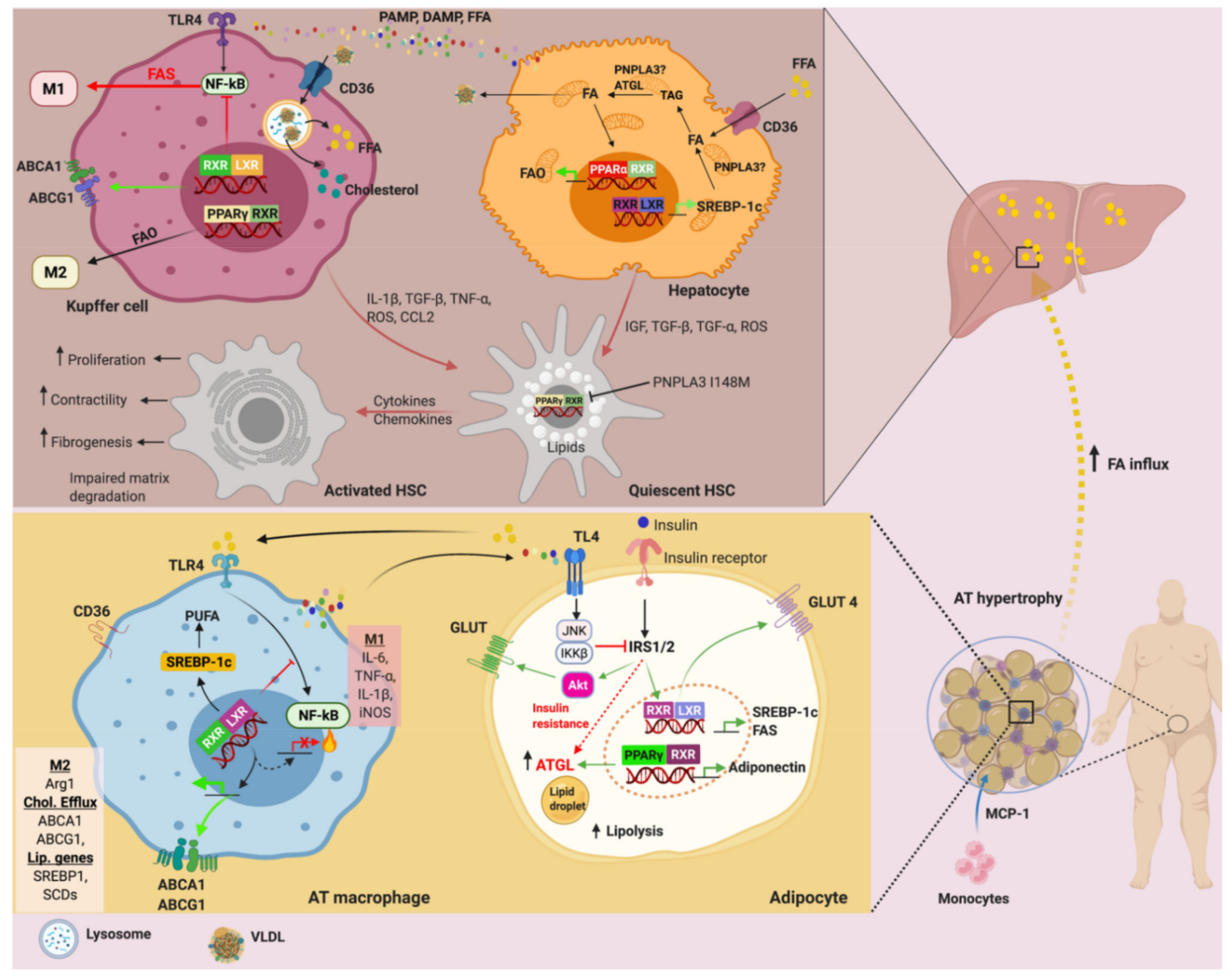
6. PPARs and LXRs in Adipose Tissue Metabolism
7. PPARs and LXRs in Adipose Tissue Inflammation
8. PNPLA3, ATGL, PPAR and LXR in NAFLD: A Brief Update
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| ADD-1 | Adipocyte differentiation and determinant factor |
| ACC | Acetyl-CoA carboxylase |
| AF-1/2 | Activating factor 1, 2 |
| ATGL | Adipose triglyceride lipase |
| BAT | Brown adipose tissue |
| CCL2 | C–C motif chemokine ligand 2 |
| C/EBP | CCAAT enhancer-binding protein |
| COX | Cyclooxygenase |
| DAG | Diacylglycerol |
| DBD | DNA-binding domain |
| DR1 | Direct repeat 1 |
| FASN | Fatty acid synthase |
| FFA | Free fatty acids |
| HSC | Hepatic stellate cells |
| HETE | Hydroxyeicosatetraenoic acid |
| HODE | Hydroxyoctadeca-9Z,11E-dienoic acids |
| HSL | Hormone-sensitive lipase |
| IκκB | Inhibitor of nuclear factor κB |
| IR | Insulin resistance |
| JNK | c-Jun N-terminal kinases |
| LBD | Ligand-binding domain |
| LPL | Lipoprotein lipase |
| LOX | Lipoxygenase |
| LTB4 | Leukotriene B4 |
| LXR | Liver X receptor |
| MGL | Monoglyceride Lipase |
| NAFLD | Non alcoholic fatty liver disease |
| NF-κB | Nuclear factor κB |
| PDG2 | Prostaglandin G2 |
| PNPLA3 | Patatin-like phospholipase domain containing 3 |
| PPAR | Peroxisome proliferator-activated receptor |
| PPRE | PPAR response element |
| PUFA | Polyunsaturated fatty acid |
| RXR | Retinoid X receptor |
| SFA | Saturated fatty acid |
| SCD-1 | Stearoyl-CoA desaturase 1 |
| SMRT SREBP-1 | Silent mediator of retinoic acid receptor and thyroid receptor Sterol regulatory element-binding protein 1 |
| TAG | Triacylglycerol |
| TLR4 | Toll-like receptor 4 |
| TGF-β | Transforming growth factor β |
| TNF-α | Tumor necrosis factor α |
| TZD | Thiazolidinediones |
| T2DM | Type 2 diabetes mellitus |
| WAT | White adipose tissue |
| 15d-PGJ2 | 15-deoxy-Δ12,14-prostaglandin J2 |
References
- Freese, J.; Klement, R.; Ruiz-Núñez, B.; Schwarz, S.; Lötzerich, H. The sedentary (r)evolution: Have we lost our metabolic flexibility? F1000Research 2018, 6, 1787. [Google Scholar] [CrossRef] [PubMed]
- Aucouturier, J.; Duché, P.; Timmons, B.W. Metabolic flexibility and obesity in children and youth. Obes. Rev. 2011, 12, e44–e53. [Google Scholar] [CrossRef] [PubMed]
- Corpeleijn, E.; Saris, W.H.; Blaak, E.E. Metabolic flexibility in the development of insulin resistance and type 2 diabetes: Effects of lifestyle. Obes. Rev. 2009, 10, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol. Rev. 2017, 179, 142–157. [Google Scholar] [CrossRef]
- Naiman, S.; Huynh, F.K.; Gil, R.; Glick, Y.; Shahar, Y.; Touitou, N.; Nahum, L.; Avivi, M.Y.; Roichman, A.; Kanfi, Y.; et al. SIRT6 Promotes Hepatic β-Oxidation via Activation of PPARα. Cell Rep. 2019, 29, 4127–4143.e4128. [Google Scholar] [CrossRef] [Green Version]
- Minnich, A.; Tian, N.; Byan, L.; Bilder, G. A potent PPARalpha agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E270–E279. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Durand, B.; Saunders, M.; Gaudon, C.; Roy, B.; Losson, R.; Chambon, P. Activation function-2 (AF-2) of retinoic acid receptor and 9-cis retinoic acid receptor—Presence of a conserved autonomous constitutive activating domain and influence of the nature of the response element on AF-2 activity. EMBO J. 1994, 13, 5370–5382. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Schoonjans, K.; Gelman, L.; Kim, J.B.; Najib, J.; Martin, G.; Fruchart, J.-C.; Briggs, M.; Spiegelman, B.M.; Auwerx, J. Regulation of Peroxisome Proliferator-Activated Receptor γ Expression by Adipocyte Differentiation and Determination Factor 1/Sterol Regulatory Element Binding Protein 1: Implications for Adipocyte Differentiation and Metabolism. Mol. Cell. Biol. 1999, 19, 5495. [Google Scholar] [CrossRef] [Green Version]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-γ-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Nielsen, R.; Grøntved, L.; Stunnenberg, H.G.; Mandrup, S. Peroxisome proliferator-activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol. Cell Biol. 2006, 26, 5698–5714. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, A.; Yamauchi, T.; Takekawa, S.; Hada, Y.; Ito, Y.; Maki, T.; Kadowaki, T. Peroxisome Proliferator–Activated Receptor (PPAR)α Activation Increases Adiponectin Receptors and Reduces Obesity-Related Inflammation in Adipose Tissue. Diabetes 2005, 54, 3358. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J.; Schoonjans, K.; Fruchart, J.-C.; Staels, B. Regulation of Triglyceride Metabolism by PPARs: Fibrates and Thiazolidinediones have Distinct Effects. J. Atheroscler. Thromb. 1996, 3, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariou, B.; Charbonnel, B.; Staels, B. Thiazolidinediones and PPARγ agonists: Time for a reassessment. Trends Endocrinol. Metab. 2012, 23, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.M.; Toubro, S.; Astrup, A. PPARgamma agonists in the treatment of type II diabetes: Is increased fatness commensurate with long-term efficacy? Int. J. Obes. 2003, 27, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Minireview: Challenges and opportunities in development of PPAR agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Caldwell, S.; Neuschwander-Tetri, B.A. Therapeutic trials in nonalcoholic steatohepatitis: Insulin sensitizers and related methodological issues. Hepatology 2010, 52, 2206–2215. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Waki, H.; Kamon, J.; Murakami, K.; Motojima, K.; Komeda, K.; Miki, H.; Kubota, N.; Terauchi, Y.; Tsuchida, A.; et al. Inhibition of RXR and PPARγ ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Investig. 2001, 108, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fujimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarhangi, N.; Sharifi, F.; Hashemian, L.; Hassani Doabsari, M.; Heshmatzad, K.; Rahbaran, M.; Jamaldini, S.H.; Aghaei Meybodi, H.R.; Hasanzad, M. PPARG (Pro12Ala) genetic variant and risk of T2DM: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12764. [Google Scholar] [CrossRef]
- Li, M.; Pascual, G.; Glass, C.K. Peroxisome Proliferator-Activated Receptor γ-Dependent Repression of the Inducible Nitric Oxide Synthase Gene. Mol. Cell. Biol. 2000, 20, 4699. [Google Scholar] [CrossRef] [Green Version]
- Helzer, K.T.; Hooper, C.; Miyamoto, S.; Alarid, E.T. Ubiquitylation of nuclear receptors: New linkages and therapeutic implications. J. Mol. Endocrinol. 2015, 54, R151–R167. [Google Scholar] [CrossRef] [Green Version]
- Jennewein, C.; Kuhn, A.M.; Schmidt, M.V.; Meilladec-Jullig, V.; Von Knethen, A.; Gonzalez, F.J.; Brüne, B. Sumoylation of peroxisome proliferator-activated receptor γ by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from κB binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 2008, 181, 5646–5652. [Google Scholar] [CrossRef] [Green Version]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXR α. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef] [Green Version]
- Beaven, S.W.; Matveyenko, A.; Wroblewski, K.; Chao, L.; Wilpitz, D.; Hsu, T.W.; Lentz, J.; Drew, B.; Hevener, A.L.; Tontonoz, P. Reciprocal regulation of hepatic and adipose lipogenesis by liver X receptors in obesity and insulin resistance. Cell Metab. 2013, 18, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, M.N.; Hong, C.; Chen, M.; Joseph, S.B.; Wilpitz, D.C.; Wang, X.; Lusis, A.J.; Collins, A.; Hseuh, W.A.; Collins, J.L.; et al. Ligand activation of LXR β reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR α and apoE. J. Clin. Investig. 2007, 117, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Breevoort, S.R.; Angdisen, J.; Fu, M.; Schmidt, D.R.; Holmstrom, S.R.; Kliewer, S.A.; Mangelsdorf, D.J.; Schulman, I.G. Liver LXRα expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Investig. 2012, 122, 1688–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korach-André, M.; Archer, A.; Gabbi, C.; Barros, R.P.; Pedrelli, M.; Steffensen, K.R.; Pettersson, A.T.; Laurencikiene, J.; Parini, P.; Gustafsson, J. Liver X receptors regulate de novo lipogenesis in a tissue-specific manner in C57BL/6 female mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E210–E222. [Google Scholar] [CrossRef] [Green Version]
- Ouvrier, A.; Cadet, R.; Vernet, P.; Laillet, B.; Chardigny, J.-M.; Lobaccaro, J.-M.A.; Drevet, J.R.; Saez, F. LXR and ABCA1 control cholesterol homeostasis in the proximal mouse epididymis in a cell-specific manner. J. Lipid Res. 2009, 50, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Kotokorpi, P.; Ellis, E.; Parini, P.; Nilsson, L.-M.; Strom, S.; Steffensen, K.R.; Gustafsson, J.-Å.; Mode, A. Physiological Differences between Human and Rat Primary Hepatocytes in Response to Liver X Receptor Activation by 3-[3-[N-(2-Chloro-3-trifluoromethylbenzyl)-(2,2-diphenylethyl)amino]propyloxy]phenylacetic Acid Hydrochloride (GW3965). Mol. Pharmacol. 2007, 72, 947–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR α. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Flynn, L.; Woodhouse, K.A. Adipose tissue engineering with cells in engineered matrices. Organogenesis 2008, 4, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Guo, Y.-Y.; Chi, Q.-S.; Zhang, X.-Y.; Liu, W.; Hao, S.-Y.; Wang, D.-H. Brown adipose tissue plays thermoregulatory role within the thermoneutral zone in Mongolian gerbils (Meriones unguiculatus). J. Therm. Biol. 2019, 81, 137–145. [Google Scholar] [CrossRef]
- Fischer-Posovszky, P.; Newell, F.S.; Wabitsch, M.; Tornqvist, H.E. Human SGBS cells—A unique tool for studies of human fat cell biology. Obes. Facts 2008, 1, 184–189. [Google Scholar] [CrossRef]
- Valli, V.; Heilmann, K.; Danesi, F.; Bordoni, A.; Gerhäuser, C. Modulation of Adipocyte Differentiation and Proadipogenic Gene Expression by Sulforaphane, Genistein, and Docosahexaenoic Acid as a First Step to Counteract Obesity. Oxidative Med. Cell. Longev. 2018, 2018, 1617202. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, K.W.; Lee, E.W.; Jang, W.S.; Seo, J.; Shin, S.; Hwang, K.A.; Song, J. Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 2014, 21, 594–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntambi, J.M.; Young-Cheul, K. Adipocyte differentiation and gene expression. J. Nutr. 2000, 130, 3122s–3126s. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR γ mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Payne, V.A.; Au, W.-S.; Lowe, C.E.; Rahman, S.M.; Friedman, J.E.; O’Rahilly, S.; Rochford, J.J. C/EBP transcription factors regulate SREBP1c gene expression during adipogenesis. Biochem. J. 2009, 425, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Q.; Tsai, J.; Tan, G.; Dalgin, G.; Hotamisligil, G.S. Interaction between GATA and the C/EBP family of transcription factors is critical in GATA-mediated suppression of adipocyte differentiation. Mol. Cell Biol. 2005, 25, 706–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajas, L.; Fruchart, J.-C.; Auwerx, J. Transcriptional control of adipogenesis. Curr. Opin. Cell Biol. 1998, 10, 165–173. [Google Scholar] [CrossRef]
- Wu, Z.; Rosen, E.D.; Brun, R.; Hauser, S.; Adelmant, G.; Troy, A.E.; McKeon, C.; Darlington, G.J.; Spiegelman, B.M. Cross-regulation of C/EBP α and PPAR γ controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol. Cell 1999, 3, 151–158. [Google Scholar] [CrossRef]
- Vernochet, C.; Peres, S.B.; Davis, K.E.; McDonald, M.E.; Qiang, L.; Wang, H.; Scherer, P.E.; Farmer, S.R. C/EBPalpha and the corepressors CtBP1 and CtBP2 regulate repression of select visceral white adipose genes during induction of the brown phenotype in white adipocytes by peroxisome proliferator-activated receptor γ agonists. Mol. Cell Biol. 2009, 29, 4714–4728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, E.; Tontonoz, P.; Spiegelman, B.M. Transdifferentiation of myoblasts by the adipogenic transcription factors PPAR γ and C/EBP α. Proc. Natl. Acad. Sci. USA 1995, 92, 9856–9860. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Zeve, D.; Seo, J.; Jo, A.Y.; Graff, J.M. Thiazolidinediones regulate adipose lineage dynamics. Cell Metab. 2011, 14, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Camp, H.S.; Whitton, A.L.; Tafuri, S.R. PPARgamma activators down-regulate the expression of PPARgamma in 3T3-L1 adipocytes. FEBS Lett. 1999, 447, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Spiegelman, B.M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev. 1996, 10, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Osuga, J.; Ishibashi, S.; Oka, T.; Yagyu, H.; Tozawa, R.; Fujimoto, A.; Shionoiri, F.; Yahagi, N.; Kraemer, F.B.; Tsutsumi, O.; et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehner, R.; Quiroga, A. Fatty Acid Handling in Mammalian Cells; Elsevier: Amsterdam, The Netherlands, 2016; pp. 149–184. [Google Scholar]
- Gruber, A.; Cornaciu, I.; Lass, A.; Schweiger, M.; Poeschl, M.; Eder, C.; Kumari, M.; Schoiswohl, G.; Wolinski, H.; Kohlwein, S.D.; et al. The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 12289–12298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef]
- Schweiger, M.; Romauch, M.; Schreiber, R.; Grabner, G.F.; Hütter, S.; Kotzbeck, P.; Benedikt, P.; Eichmann, T.O.; Yamada, S.; Knittelfelder, O.; et al. Pharmacological inhibition of adipose triglyceride lipase corrects high-fat diet-induced insulin resistance and hepatosteatosis in mice. Nat. Commun. 2017, 8, 14859. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, N.; Takahara, S.; Matsumura, N.; Kim, T.T.; Ferdaoussi, M.; Migglautsch, A.K.; Zechner, R.; Breinbauer, R.; Kershaw, E.E.; Dyck, J.R.B. Atglistatin ameliorates functional decline in heart failure via adipocyte-specific inhibition of adipose triglyceride lipase. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H879–H884. [Google Scholar] [CrossRef] [PubMed]
- Kralisch, S.; Klein, J.; Lossner, U.; Bluher, M.; Paschke, R.; Stumvoll, M.; Fasshauer, M. Isoproterenol, TNFalpha, and insulin downregulate adipose triglyceride lipase in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2005, 240, 43–49. [Google Scholar] [CrossRef]
- Roy, D.; Farabaugh, K.T.; Wu, J.; Charrier, A.; Smas, C.; Hatzoglou, M.; Thirumurugan, K.; Buchner, D.A. Coordinated transcriptional control of adipocyte triglyceride lipase (Atgl) by transcription factors Sp1 and peroxisome proliferator-activated receptor γ (PPARγ) during adipocyte differentiation. J. Biol. Chem. 2017, 292, 14827–14835. [Google Scholar] [CrossRef] [Green Version]
- Jha, P.; Claudel, T.; Baghdasaryan, A.; Mueller, M.; Halilbasic, E.; Das, S.K.; Lass, A.; Zimmermann, R.; Zechner, R.; Hoefler, G.; et al. Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology 2014, 59, 858–869. [Google Scholar] [CrossRef]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011, 53, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.L.; Zhang, X.; Saarinen, A.M.; Schoiswohl, G.; Kershaw, E.E.; Zechner, R.; Liu, J. Liver X receptor α mediates hepatic triglyceride accumulation through upregulation of G0/G1 Switch Gene 2 expression. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oosterveer, M.H.; van Dijk, T.H.; Grefhorst, A.; Bloks, V.W.; Havinga, R.; Kuipers, F.; Reijngoud, D.J. Lxralpha deficiency hampers the hepatic adaptive response to fasting in mice. J. Biol. Chem. 2008, 283, 25437–25445. [Google Scholar] [CrossRef] [Green Version]
- Fredrikson, G.; Strålfors, P.; Nilsson, N.O.; Belfrage, P. Hormone-sensitive lipase of rat adipose tissue. Purification and some properties. J. Biol. Chem. 1981, 256, 6311–6320. [Google Scholar] [CrossRef]
- Tsiloulis, T.; Watt, M.J. Exercise and the Regulation of Adipose Tissue Metabolism. Prog. Mol. Biol. Transl. Sci. 2015, 135, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, W.T.; Laplante, M.; Berthiaume, M.; Gélinas, Y.; Deshaies, Y. PPARgamma agonism increases rat adipose tissue lipolysis, expression of glyceride lipases, and the response of lipolysis to hormonal control. Diabetologia 2006, 49, 2427–2436. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.F.; Purushotham, A.; Wendel, A.A.; Koba, K.; DeIuliis, J.; Lee, K.; Belury, M.A. Regulation of adipose triglyceride lipase by rosiglitazone. Diabetes Obes. Metab. 2009, 11, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Xia, B.; Cai, G.H.; Yang, H.; Wang, S.P.; Mitchell, G.A.; Wu, J.W. Adipose tissue deficiency of hormone-sensitive lipase causes fatty liver in mice. PLoS Genet. 2017, 13, e1007110. [Google Scholar] [CrossRef] [Green Version]
- Roduit, R.; Masiello, P.; Wang, S.P.; Li, H.; Mitchell, G.A.; Prentki, M. A role for hormone-sensitive lipase in glucose-stimulated insulin secretion: A study in hormone-sensitive lipase-deficient mice. Diabetes 2001, 50, 1970–1975. [Google Scholar] [CrossRef] [Green Version]
- Peyot, M.-L.; Nolan, C.J.; Soni, K.; Joly, E.; Lussier, R.; Corkey, B.E.; Wang, S.P.; Mitchell, G.A.; Prentki, M. Hormone-Sensitive Lipase Has a Role in Lipid Signaling for Insulin Secretion but Is Nonessential for the Incretin Action of Glucagon-Like Peptide 1. Diabetes 2004, 53, 1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costabile, G.; Annuzzi, G.; Di Marino, L.; De Natale, C.; Giacco, R.; Bozzetto, L.; Cipriano, P.; Santangelo, C.; Masella, R.; Rivellese, A.A. Fasting and post-prandial adipose tissue lipoprotein lipase and hormone-sensitive lipase in obesity and Type 2 diabetes. J. Endocrinol. Investig. 2011, 34, e110–e114. [Google Scholar] [CrossRef] [PubMed]
- Chanda, P.K.; Gao, Y.; Mark, L.; Btesh, J.; Strassle, B.W.; Lu, P.; Piesla, M.J.; Zhang, M.Y.; Bingham, B.; Uveges, A.; et al. Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol. Pharm. 2010, 78, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Tardelli, M. Monoacylglycerol lipase reprograms lipid precursors signaling in liver disease. World J. Gastroenterol. 2020, 26, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Mulvihill, M.M.; Mukhopadhyay, P.; Xu, H.; Erdélyi, K.; Hao, E.; Holovac, E.; Haskó, G.; Cravatt, B.F.; Nomura, D.K.; et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 2013, 144, 808–817.e815. [Google Scholar] [CrossRef] [Green Version]
- Taschler, U.; Radner, F.P.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Sanderson, L.M.; Matilainen, M.; Stienstra, R.; Carlberg, C.; de Groot, P.J.; Müller, M.; Kersten, S. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res. 2007, 2007, 26839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.D.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.F.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292. [Google Scholar] [CrossRef]
- Strauss, J.G.; Frank, S.; Kratky, D.; Hämmerle, G.; Hrzenjak, A.; Knipping, G.; von Eckardstein, A.; Kostner, G.M.; Zechner, R. Adenovirus-mediated rescue of lipoprotein lipase-deficient mice. Lipolysis of triglyceride-rich lipoproteins is essential for high density lipoprotein maturation in mice. J. Biol. Chem. 2001, 276, 36083–36090. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Repa, J.J.; Gauthier, K.; Mangelsdorf, D.J. Regulation of lipoprotein lipase by the oxysterol receptors, LXRalpha and LXRbeta. J. Biol. Chem. 2001, 276, 43018–43024. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Tang, J.; Hu, X.; Bao, P.; Pan, J.; Chen, Z.; Xian, J. MiR-27b Impairs Adipocyte Differentiation of Human Adipose Tissue-Derived Mesenchymal Stem Cells by Targeting LPL. Cell Physiol. Biochem. 2018, 47, 545–555. [Google Scholar] [CrossRef]
- Laplante, M.; Sell, H.; MacNaul, K.L.; Richard, D.; Berger, J.P.; Deshaies, Y. PPAR-γ activation mediates adipose depot-specific effects on gene expression and lipoprotein lipase activity: Mechanisms for modulation of postprandial lipemia and differential adipose accretion. Diabetes 2003, 52, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, K.; Lopez, C.; Donovan, D.; Ngai, C.; Fontanez, N.; Bensadoun, A.; Fruchart-Najib, J.; Holleran, S.; Cohn, J.S.; Ramakrishnan, R.; et al. Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J. Clin. Investig. 2005, 115, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, I.; Salomon, H.; Després, J.P. Contribution of apo CIII reduction to the greater effect of 12-week micronized fenofibrate than atorvastatin therapy on triglyceride levels and LDL size in dyslipidemic patients. Ann. Med. 2003, 35, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.M.; Parra, H.J.; Camare, R.; Luc, G.; Ziegler, O.; Dachet, C.; Bruckert, E.; Douste-Blazy, P.; Drouin, P.; Jacotot, B.; et al. A multicenter comparison of the effects of simvastatin and fenofibrate therapy in severe primary hypercholesterolemia, with particular emphasis on lipoproteins defined by their apolipoprotein composition. Metabolism 1992, 41, 498–503. [Google Scholar] [CrossRef]
- Gervois, P.; Chopin-Delannoy, S.; Fadel, A.; Dubois, G.; Kosykh, V.; Fruchart, J.-C.; Najïb, J.; Laudet, V.; Staels, B. Fibrates Increase Human REV-ERBα Expression in Liver via a Novel Peroxisome Proliferator-Activated Receptor Response Element. Mol. Endocrinol. 1999, 13, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, R.; Bishara-Shieban, J.; Bar-Tana, J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J. Biol. Chem. 1995, 270, 13470–13475. [Google Scholar] [CrossRef] [Green Version]
- Schultze, A.E.; Alborn, W.E.; Newton, R.K.; Konrad, R.J. Administration of a PPARalpha agonist increases serum apolipoprotein A-V levels and the apolipoprotein A-V/apolipoprotein C-III ratio. J. Lipid Res. 2005, 46, 1591–1595. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Ruuska, S.E.; Shaw, N.S.; Dong, D.; Noy, N. Ligand selectivity of the peroxisome proliferator-activated receptor α. Biochemistry 1999, 38, 185–190. [Google Scholar] [CrossRef]
- Zuo, X.; Wu, Y.; Morris, J.S.; Stimmel, J.B.; Leesnitzer, L.M.; Fischer, S.M.; Lippman, S.M.; Shureiqi, I. Oxidative metabolism of linoleic acid modulates PPAR-β/delta suppression of PPAR-γ activity. Oncogene 2006, 25, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-W.; Chien, Y.-S.; Chen, Y.-J.; Ajuwon, K.M.; Mersmann, H.M.; Ding, S.-T. Role of n-3 Polyunsaturated Fatty Acids in Ameliorating the Obesity-Induced Metabolic Syndrome in Animal Models and Humans. Int. J. Mol. Sci. 2016, 17, 1689. [Google Scholar] [CrossRef]
- Calder, P.C.; Grimble, R.F. Polyunsaturated fatty acids, inflammation and immunity. Eur. J. Clin. Nutr. 2002, 56, S14–S19. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Su, C.W.; Kang, J.X. Endogenous Omega-3 Polyunsaturated Fatty Acids Reduce the Number and Differentiation of White Adipocyte Progenitors in Mice. Obesity 2020, 28, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessler, A.M.; Kaur, N.; Palta, J.P.; Ntambi, J.M. Regulation of stearoyl-CoA desaturase 1 mRNA stability by polyunsaturated fatty acids in 3T3-L1 adipocytes. J. Biol. Chem. 1996, 271, 29854–29858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Tu, H.; Shan, B.; Luk, A.; DeBose-Boyd, R.A.; Bashmakov, Y.; Goldstein, J.L.; Brown, M.S. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci. USA 2001, 98, 6027–6032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, T.; Shimano, H.; Yahagi, N.; Ide, T.; Amemiya-Kudo, M.; Matsuzaka, T.; Nakakuki, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem. 2002, 277, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.D.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARγ Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735. [Google Scholar] [CrossRef] [Green Version]
- Araki, E.; Yamashita, S.; Arai, H.; Yokote, K.; Satoh, J.; Inoguchi, T.; Nakamura, J.; Maegawa, H.; Yoshioka, N.; Tanizawa, Y.; et al. Effects of Pemafibrate, a Novel Selective PPARα Modulator, on Lipid and Glucose Metabolism in Patients With Type 2 Diabetes and Hypertriglyceridemia: A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Diabetes Care 2018, 41, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Bhat, Z.R.; Bhat, S.; Kumar, R.; Kumar, R.; Tikoo, K.; Gupta, J.; Khurana, N.; Kaur, J.; Khatik, G.L. Synthesis and evaluation of new 1,2,4-oxadiazole based trans- acrylic acid derivatives as potential PPAR-α/γ dual agonist. Bioorganic Chem. 2020, 100, 103867. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Wright, R.S.; Farkouh, M.; Plutzky, J. Modulating peroxisome proliferator-activated receptors for therapeutic benefit? Biology, clinical experience, and future prospects. Am. Heart J. 2012, 164, 672–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.; Gupta, R.A.; Ma, W.G.; Paria, B.C.; Moller, D.E.; Morrow, J.D.; DuBois, R.N.; Trzaskos, J.M.; Dey, S.K. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARdelta. Genes Dev. 1999, 13, 1561–1574. [Google Scholar] [CrossRef]
- Coleman, J.D.; Prabhu, K.S.; Thompson, J.T.; Reddy, P.S.; Peters, J.M.; Peterson, B.R.; Reddy, C.C.; Vanden Heuvel, J.P. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-β/delta (PPARbeta/delta). Free Radic. Biol. Med. 2007, 42, 1155–1164. [Google Scholar] [CrossRef] [Green Version]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.Z.; Fischer, S.M.; Lippman, S.M. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968–9973. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.; Leibowitz, M.D.; Doebber, T.W.; Elbrecht, A.; Zhang, B.; Zhou, G.; Biswas, C.; Cullinan, C.A.; Hayes, N.S.; Li, Y.; et al. Novel peroxisome proliferator-activated receptor (PPAR) γ and PPARdelta ligands produce distinct biological effects. J. Biol. Chem. 1999, 274, 6718–6725. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Gunn, N.T.; Khazanchi, A.; Guy, C.; Brunt, E.M.; Moussa, S.; Baum, S.; Frias, J.; Trotter, J.; Lazas, D.; et al. A 52-Week Multi-Center Double-Blind Randomized Phase 2 Study of Seladelpar, a potent and selective peroxisome proliferator-activated receptor delta (PPAR-delta) agonist, in Patients with Nonalcoholic Steatohepatitis (NASH). Hepatology 2020, 72, 1. [Google Scholar]
- Romanowska, M.; Reilly, L.; Palmer, C.N.; Gustafsson, M.C.; Foerster, J. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PLoS ONE 2010, 5, e9701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Reginato, M.J.; Krakow, S.L.; Bailey, S.T.; Lazar, M.A. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J. Biol. Chem. 1998, 273, 1855–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Murakawa, Y.; Okada, K.; Horikoshi, H.; Yokoyama, J.; Tajima, N.; Ikeda, Y. Effect of troglitazone on body fat distribution in type 2 diabetic patients. Diabetes Care 1999, 22, 908–912. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Kendall, D.M.; Deeg, M.A.; Buse, J.B.; Zagar, A.J.; Pinaire, J.A.; Tan, M.H.; Khan, M.A.; Perez, A.T.; Jacober, S.J. A Comparison of Lipid and Glycemic Effects of Pioglitazone and Rosiglitazone in Patients With Type 2 Diabetes and Dyslipidemia. Diabetes Care 2005, 28, 1547. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Kim, D.M.; Woo, J.T.; Jang, H.C.; Chung, C.H.; Ko, K.S.; Park, J.H.; Park, Y.S.; Kim, S.J.; Choi, D.S. Efficacy and safety of lobeglitazone monotherapy in patients with type 2 diabetes mellitus over 24-weeks: A multicenter, randomized, double-blind, parallel-group, placebo controlled trial. PLoS ONE 2014, 9, e92843. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Sebo, Z.L.; Rendina-Ruedy, E.; Ables, G.P.; Lindskog, D.M.; Rodeheffer, M.S.; Fazeli, P.K.; Horowitz, M.C. Bone Marrow Adiposity: Basic and Clinical Implications. Endocr. Rev. 2019, 40, 1187–1206. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.S.; Depaoli, A.M. Selective peroxisome proliferator-activated receptor γ (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 2010, 91, 267s–272s. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, K.; Hwang, B.J.; Choi, J.H.; Dewi, R.; Ou, L.; McLenithan, J.; Twaddel, W.; Pozharski, E.; Stock, J.; Girnun, G.D. N-Acetylfarnesylcysteine is a novel class of peroxisome proliferator-activated receptor γ ligand with partial and full agonist activity in vitro and in vivo. J. Biol. Chem. 2011, 286, 41626–41635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, S.; Hines, G.V.; Lozada-Fernandez, V.V.; de Jesus Piva, C.; Kaliappan, A.; Rider, S.D., Jr.; Hostetler, H.A. Fatty acid binding profile of the liver X receptor α. J. Lipid Res. 2017, 58, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, A.; Xu, J.; Jerks, E.; Mangelsdorf, D.J.; Jump, D.B. Fatty acid regulation of liver X receptors (LXR) and peroxisome proliferator-activated receptor α (PPARalpha) in HEK293 cells. J. Biol. Chem. 2002, 277, 39243–39250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Shionoiri, F.; Ohashi, K.; Osuga, J.; et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem. 1999, 274, 35840–35844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Bu, L.; Ma, Y.; Liu, D. Concurrent Activation of Liver X Receptor and Peroxisome Proliferator-Activated Receptor α Exacerbates Hepatic Steatosis in High Fat Diet-Induced Obese Mice. PLoS ONE 2013, 8, e65641. [Google Scholar] [CrossRef] [Green Version]
- Laffitte, B.A.; Chao, L.C.; Li, J.; Walczak, R.; Hummasti, S.; Joseph, S.B.; Castrillo, A.; Wilpitz, D.C.; Mangelsdorf, D.J.; Collins, J.L.; et al. Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc. Natl. Acad. Sci. USA 2003, 100, 5419–5424. [Google Scholar] [CrossRef] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Okuno, A.; Tamemoto, H.; Tobe, K.; Ueki, K.; Mori, Y.; Iwamoto, K.; Umesono, K.; Akanuma, Y.; Fujiwara, T.; Horikoshi, H.; et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J. Clin. Investig. 1998, 101, 1354–1361. [Google Scholar] [CrossRef]
- Kissebah, A.H.; Vydelingum, N.; Murray, R.; Evans, D.J.; Hartz, A.J.; Kalkhoff, R.K.; Adams, P.W. Relation of body fat distribution to metabolic complications of obesity. J. Clin. Endocrinol. Metab. 1982, 54, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Salans, L.B.; Knittle, J.L.; Hirsch, J. The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J. Clin. Investig. 1968, 47, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Sherman, A.; Tsao, P.; Gonzalez, O.; Yee, G.; Lamendola, C.; Reaven, G.M.; Cushman, S.W. Enhanced proportion of small adipose cells in insulin-resistant vs insulin-sensitive obese individuals implicates impaired adipogenesis. Diabetologia 2007, 50, 1707–1715. [Google Scholar] [CrossRef] [Green Version]
- Frohnert, B.; Hui, T.Y.; Bernlohr, D. Identification of a functional peroxisome proliferator-responsive element in the murine fatty acid transport protein gene. J. Biol. Chem. 1999, 274, 3970–3977. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-B.; Ciaraldi, T.P.; Kong, A.; Kim, D.; Chu, N.; Mohideen, P.; Mudaliar, S.; Henry, R.R.; Kahn, B.B. Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110[β] protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 2002, 51, 443. [Google Scholar] [CrossRef] [Green Version]
- Guri, A.J.; Hontecillas, R.; Ferrer, G.; Casagran, O.; Wankhade, U.; Noble, A.M.; Eizirik, D.L.; Ortis, F.; Cnop, M.; Liu, D.; et al. Loss of PPARγ in immune cells impairs the ability of abscisic acid to improve insulin sensitivity by suppressing monocyte chemoattractant protein-1 expression and macrophage infiltration into white adipose tissue. J. Nutr. Biochem. 2008, 19, 216–228. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, M.; Lo Verme, J.; Fu, J.; Oveisi, F.; Blázquez, C.; Piomelli, D. Oleoylethanolamide Stimulates Lipolysis by Activating the Nuclear Receptor Peroxisome Proliferator-activated Receptor α (PPAR-α). J. Biol. Chem. 2004, 279, 27849–27854. [Google Scholar] [CrossRef] [Green Version]
- Guerre-Millo, M.; Gervois, P.; Raspé, E.; Madsen, L.; Poulain, P.; Derudas, B.; Herbert, J.M.; Winegar, D.A.; Willson, T.M.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor α activators improve insulin sensitivity and reduce adiposity. J. Biol. Chem. 2000, 275, 16638–16642. [Google Scholar] [CrossRef] [Green Version]
- Bastie, C.; Luquet, S.; Holst, D.; Jehl-Pietri, C.; Grimaldi, P. Alterations of peroxisome proliferator-activated receptor δ activity affect fatty acid-controlled adipose differentiation. J. Biol. Chem. 2001, 275, 38768–38773. [Google Scholar] [CrossRef] [Green Version]
- Barroso, E.; Rodríguez-Rodríguez, R.; Chacón, M.R.; Maymó-Masip, E.; Ferrer, L.; Salvadó, L.; Salmerón, E.; Wabistch, M.; Palomer, X.; Vendrell, J.; et al. PPARβ/δ ameliorates fructose-induced insulin resistance in adipocytes by preventing Nrf2 activation. Biochim. Biophys. Acta 2015, 1852, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.B.; Moon, H.M.; Kim, W.S.; Lee, Y.S.; Jeong, H.W.; Yoo, E.J.; Ham, J.; Kang, H.; Park, M.G.; Steffensen, K.R.; et al. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor γ expression. Mol. Cell Biol. 2004, 24, 3430–3444. [Google Scholar] [CrossRef] [Green Version]
- Laffitte, B.A.; Joseph, S.B.; Walczak, R.; Pei, L.; Wilpitz, D.C.; Collins, J.L.; Tontonoz, P. Autoregulation of the human liver X receptor α promoter. Mol. Cell Biol. 2001, 21, 7558–7568. [Google Scholar] [CrossRef] [Green Version]
- Steffensen, K.R.; Schuster, G.U.; Parini, P.; Holter, E.; Sadek, C.M.; Cassel, T.; Eskild, W.; Gustafsson, J.A. Different regulation of the LXRalpha promoter activity by isoforms of CCAAT/enhancer-binding proteins. Biochem. Biophys. Res. Commun. 2002, 293, 1333–1340. [Google Scholar] [CrossRef]
- Hammarstedt, A.; Rotter Sopasakis, V.; Gogg, S.; Jansson, P.A.; Smith, U. Improved insulin sensitivity and adipose tissue dysregulation after short-term treatment with pioglitazone in non-diabetic, insulin-resistant subjects. Diabetologia 2005, 48, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Gerin, I.; Dolinsky, V.W.; Shackman, J.G.; Kennedy, R.T.; Chiang, S.H.; Burant, C.F.; Steffensen, K.R.; Gustafsson, J.Å.; MacDougald, O.A. LXRβ is required for adipocyte growth, glucose homeostasis, and β cell function. J. Biol. Chem. 2005, 280, 23024–23031. [Google Scholar] [CrossRef] [Green Version]
- Keuper, M.; Blüher, M.; Schön, M.R.; Möller, P.; Dzyakanchuk, A.; Amrein, K.; Debatin, K.-M.; Wabitsch, M.; Fischer-Posovszky, P. An inflammatory micro-environment promotes human adipocyte apoptosis. Mol. Cell. Endocrinol. 2011, 339, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Hellmann, J.; Tang, Y.; Kosuri, M.; Bhatnagar, A.; Spite, M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 2011, 25, 2399–2407. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Cox, N.; Geissmann, F. Macrophage ontogeny in the control of adipose tissue biology. Curr. Opin. Immunol. 2020, 62, 1–8. [Google Scholar] [CrossRef]
- McLaughlin, T.; Ackerman, S.E.; Shen, L.; Engleman, E. Role of innate and adaptive immunity in obesity-associated metabolic disease. J. Clin. Investig. 2017, 127, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [Green Version]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, B.; Nagy, G.; Czimmerer, Z.; Horvath, A.; Hammers, D.W.; Cuaranta-Monroy, I.; Poliska, S.; Tzerpos, P.; Kolostyak, Z.; Hays, T.T.; et al. The Nuclear Receptor PPARγ Controls Progressive Macrophage Polarization as a Ligand-Insensitive Epigenomic Ratchet of Transcriptional Memory. Immunity 2018, 49, 615–626.e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Leopold Wager, C.M.; Arnett, E.; Schlesinger, L.S. Macrophage nuclear receptors: Emerging key players in infectious diseases. PLoS Pathog. 2019, 15, e1007585. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef]
- Sanderson, L.M.; Boekschoten, M.V.; Desvergne, B.; Müller, M.; Kersten, S. Transcriptional profiling reveals divergent roles of PPARalpha and PPARbeta/delta in regulation of gene expression in mouse liver. Physiol Genom. 2010, 41, 42–52. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Choi, R.; Kim, H.M.; Cho, E.J.; Kim, B.H.; Choi, Y.S.; Naowaboot, J.; Lee, E.Y.; Yang, Y.C.; Shin, J.Y.; et al. Peroxisome proliferator-activated receptor δ agonist attenuates hepatic steatosis by anti-inflammatory mechanism. Exp. Mol. Med. 2012, 44, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 2015, 4, e08009. [Google Scholar] [CrossRef]
- Bi, X.; Song, J.; Gao, J.; Zhao, J.; Wang, M.; Scipione, C.A.; Koschinsky, M.L.; Wang, Z.V.; Xu, S.; Fu, G. Activation of liver X receptor attenuates lysophosphatidylcholine-induced IL-8 expression in endothelial cells via the NF-κB pathway and SUMOylation. J. Cell Mol. Med. 2016, 20, 2249–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venteclef, N.; Jakobsson, T.; Ehrlund, A.; Damdimopoulos, A.; Mikkonen, L.; Ellis, E.; Nilsson, L.-M.; Parini, P.; Jänne, O.A.; Gustafsson, J.-A.; et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010, 24, 381–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Spann, N.J.; Kaikkonen, M.U.; Lu, M.; Oh, D.Y.; Fox, J.N.; Bandyopadhyay, G.; Talukdar, S.; Xu, J.; Lagakos, W.S.; et al. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell 2013, 155, 200–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castrillo, A.; Joseph, S.B.; Vaidya, S.A.; Haberland, M.; Fogelman, A.M.; Cheng, G.; Tontonoz, P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol. Cell 2003, 12, 805–816. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.N.; Uribe, M.; Ponciano-Rodriguez, G.; Medina-Santillan, R.; Mendez-Sanchez, N. New insights into the pathophysiology of nonalcoholic fatty liver disease. Ann. Hepatol. 2009, 8 (Suppl. 1), S9–S17. [Google Scholar] [CrossRef]
- Abrams, G.A.; Kunde, S.S.; Lazenby, A.J.; Clements, R.H. Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology 2004, 40, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Gastaldelli, A.; Svegliati Baroni, G.; Tell, G.; Tiribelli, C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 2008, 14, 72–81. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving Concepts in the Pathogenesis of NASH: Beyond Steatosis and Inflammation. Int. J. Mol. Sci. 2014, 15, 8591. [Google Scholar] [CrossRef]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Moschen, A.R. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology 2021, 73, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid Res. 2009, 50, 2111–2116. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.C.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Kumari, M.; Schoiswohl, G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A.; et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Basantani, M.K.; Sitnick, M.T.; Cai, L.; Brenner, D.S.; Gardner, N.P.; Li, J.Z.; Schoiswohl, G.; Yang, K.; Kumari, M.; Gross, R.W.; et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J. Lipid Res. 2011, 52, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Chang, B.; Li, L.; Chan, L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology 2010, 52, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Huang, Y.; Karaman, R.; Ivanova, P.T.; Brown, H.A.; Roddy, T.; Castro-Perez, J.; Cohen, J.C.; Hobbs, H.H. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J. Clin. Investig. 2012, 122, 4130–4144. [Google Scholar] [CrossRef] [Green Version]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C., Jr.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, P.-J.; Chiou, H.-Y.C.; Jiang, H.-J.; Lee, M.-Y.; Hsieh, T.-J.; Kuo, K.-K. Pioglitazone Enhances Cytosolic Lipolysis, β-oxidation and Autophagy to Ameliorate Hepatic Steatosis. Sci. Rep. 2017, 7, 9030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi-Suzuki, M.; Bril, F.; Kalavalapalli, S.; Cusi, K.; Frye, R.F. Concentration-dependent response to pioglitazone in nonalcoholic steatohepatitis. Aliment Pharm. Ther. 2017, 46, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheen, A.J. Pharmacokinetic interactions with thiazolidinediones. Clin Pharm. 2007, 46, 1–12. [Google Scholar] [CrossRef]
- Kawaguchi-Suzuki, M.; Cusi, K.; Bril, F.; Gong, Y.; Langaee, T.; Frye, R.F. A Genetic Score Associates With Pioglitazone Response in Patients With Non-alcoholic Steatohepatitis. Front. Pharmacol. 2018, 9, 752. [Google Scholar] [CrossRef]
- Panunzi, S.; Maltese, S.; Verrastro, O.; Labbate, L.; De Gaetano, A.; Pompili, M.; Capristo, E.; Bornstein, S.R.; Mingrone, G. Pioglitazone and bariatric surgery are the most effective treatments for non-alcoholic steatohepatitis: A hierarchical network meta-analysis. Diabetes Obes. Metab. 2020. [Google Scholar] [CrossRef]
- Ip, E.; Farrell, G.C.; Robertson, G.; Hall, P.; Kirsch, R.; Leclercq, I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003, 38, 123–132. [Google Scholar] [CrossRef]
- Westerouen Van Meeteren, M.J.; Drenth, J.P.H.; Tjwa, E.T.T.L. Elafibranor: A potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 117–123. [Google Scholar] [CrossRef]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR α/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Ratziu, V.; Bedossa, P.; Dufour, J.F.; Krugar, F.; Schattenberg, J.M.; Francque, S.M.; Arrese, M.; George, J.; Bugianesi, E.; et al. RESOLVE-IT Phase 3 of Elafibranor in NASH: Final Results of the Week 72 Interim Surrogate Efficacy Analysis. Hepatology 2020, 72. [Google Scholar]
- Kumar, D.P.; Caffrey, R.; Marioneaux, J.; Santhekadur, P.K.; Bhat, M.; Alonso, C.; Koduru, S.V.; Philip, B.; Jain, M.R.; Giri, S.R.; et al. The PPAR α/γ Agonist Saroglitazar Improves Insulin Resistance and Steatohepatitis in a Diet Induced Animal Model of Nonalcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 9330. [Google Scholar] [CrossRef]
- Nageeb, M.M.; Khatab, M.I.; Abdel-sameea, A.A.; Teleb, N.A. Adelmidrol protects against non-alcoholic steatohepatitis in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 777–784. [Google Scholar] [CrossRef]
- Francque, S.; Bedossa, P.; Ratziu, V.; Anstee, Q.; Bugianesi, E.; Sanyal, A.; Loomba, R.; Harrison, S.A.; Balabanska, R.v.; Mateva, L.; et al. The PanPPAR agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic NASH: Results of the NATIVE Phase 2b trial. Hepatology 2020, 72. [Google Scholar]
- Hao, Y.; Wang, X.; Zhang, F.; Wang, M.; Wang, Y.; Wang, H.; Du, Y.; Wang, T.; Fu, F.; Gao, Z.; et al. Inhibition of notch enhances the anti-atherosclerotic effects of LXR agonists while reducing fatty liver development in ApoE-deficient mice. Toxicol. Appl. Pharm. 2020, 406, 115211. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.N.; Wang, C.C.N.; Chang, H.Y.; Chu, F.Y.; Hsu, Y.A.; Cheng, W.K.; Ma, W.C.; Chen, C.J.; Wan, L.; Lim, Y.P. Ursolic Acid, a Novel Liver X Receptor α (LXRα) Antagonist Inhibiting Ligand-Induced Nonalcoholic Fatty Liver and Drug-Induced Lipogenesis. J. Agric. Food Chem. 2018, 66, 11647–11662. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, M.; Zhang, B.; Zhao, J.; Feng, Q.; Peng, J.; Hu, Y.; Zhao, Y. Biological mechanisms and related natural modulators of liver X receptor in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2019, 113, 108778. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef]
- Liu, Y.; Han, X.; Bian, Z.; Peng, Y.; You, Z.; Wang, Q.; Chen, X.; Qiu, D.; Ma, X. Activation of liver X receptors attenuates endotoxin-induced liver injury in mice with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2012, 57, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Kaluba, B.; Jiang, X.L.; Chang, S.; Tang, X.F.; Mao, L.F.; Zhang, Z.P.; Huang, F.Z. Liver X Receptor Inverse Agonist SR9243 Suppresses Nonalcoholic Steatohepatitis Intrahepatic Inflammation and Fibrosis. Biomed. Res. Int. 2018, 2018, 8071093. [Google Scholar] [CrossRef] [Green Version]
- Griffett, K.; Solt, L.A.; El-Gendy Bel, D.; Kamenecka, T.M.; Burris, T.P. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol. 2013, 8, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Griffett, K.; Welch, R.D.; Flaveny, C.A.; Kolar, G.R.; Neuschwander-Tetri, B.A.; Burris, T.P. The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol. Metab. 2015, 4, 353–357. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Nuclear Receptors | Tissue Distribution | Endogenous Ligands | Synthetic Ligand | Metabolic Function |
|---|---|---|---|---|
| PPARα | Liver, heart, kidney, muscle | Saturated and unsaturated fatty acids, eicosanoids, 8(S)-HETE, and leukotriene B4 | Fenofibrate Clofibrate Gemfibrozil Wy-14643 | Fatty acid oxidation, lipoprotein metabolism, inflammatory control |
| PPARγ | Adipocytes, liver, kidney, macrophages | Saturated and unsaturated fatty acids, 15d-PGJ2, 15-HETE, 9-HODE, and 13-HODE | Rosiglitazone Pioglitazone Troglitazone Ciglitazone Farglitazar | Fatty acid storage, lipoprotein and glucose metabolism, inflammatory control |
| PPARβ/δ | Ubiquitously expressed, high in heart, muscle, liver | Saturated and unsaturated fatty acids, and 8(S)-HETE | GW-501516 GW0742 L-165041 | Fatty acid oxidation, lipoprotein and glucose metabolism, inflammatory control |
| LXRα/β | Liver, adipose tissues, macrophages, ubiquitous (LXRβ) | Oxysterols | T-0901317 GW3695 | Fatty acid and cholesterol metabolism, glucose metabolism, inflammatory control |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixon, E.D.; Nardo, A.D.; Claudel, T.; Trauner, M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes 2021, 12, 645. https://doi.org/10.3390/genes12050645
Dixon ED, Nardo AD, Claudel T, Trauner M. The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes. 2021; 12(5):645. https://doi.org/10.3390/genes12050645
Chicago/Turabian StyleDixon, Emmanuel D., Alexander D. Nardo, Thierry Claudel, and Michael Trauner. 2021. "The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD" Genes 12, no. 5: 645. https://doi.org/10.3390/genes12050645
APA StyleDixon, E. D., Nardo, A. D., Claudel, T., & Trauner, M. (2021). The Role of Lipid Sensing Nuclear Receptors (PPARs and LXR) and Metabolic Lipases in Obesity, Diabetes and NAFLD. Genes, 12(5), 645. https://doi.org/10.3390/genes12050645