
Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives

 ,
,  and
and
Abstract
:1. Introduction
2. Can the Current Understanding of the CF Microbiota Shed Light on Genotype–Phenotype Associations?
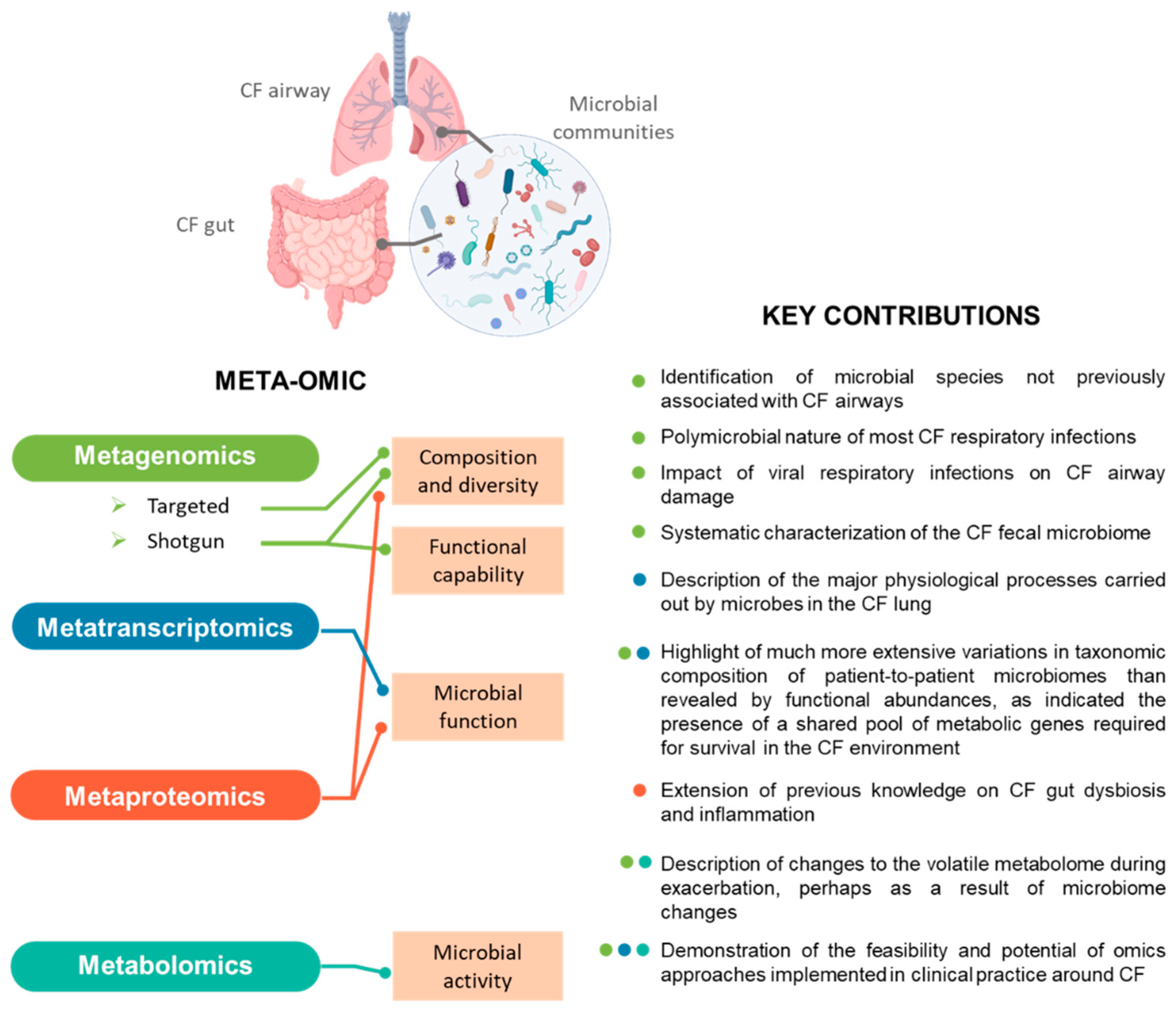
3. Meta-Omics in CF Microbiota Research
3.1. Metagenomics Profiling of CF Microbiota
3.2. Metatranscriptomics Profiling of CF Microbiota
3.3. Metabolomics Profiling of CF Microbiota
4. (Meta) Proteomics-Based Approaches Applied to CF Microbiota: Where Are We Today?
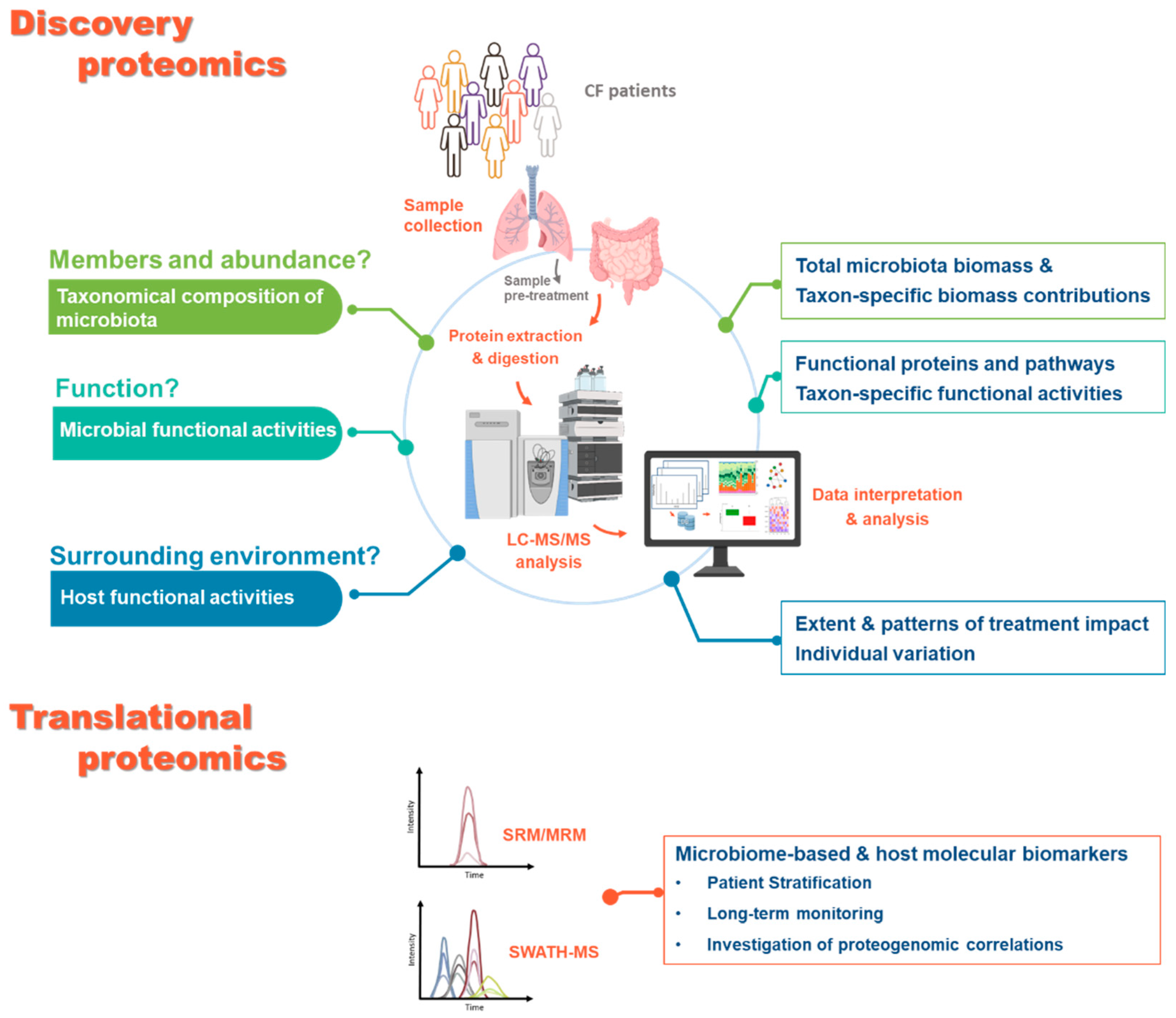
4.1. Metaproteomics Profiling of CF Microbiota
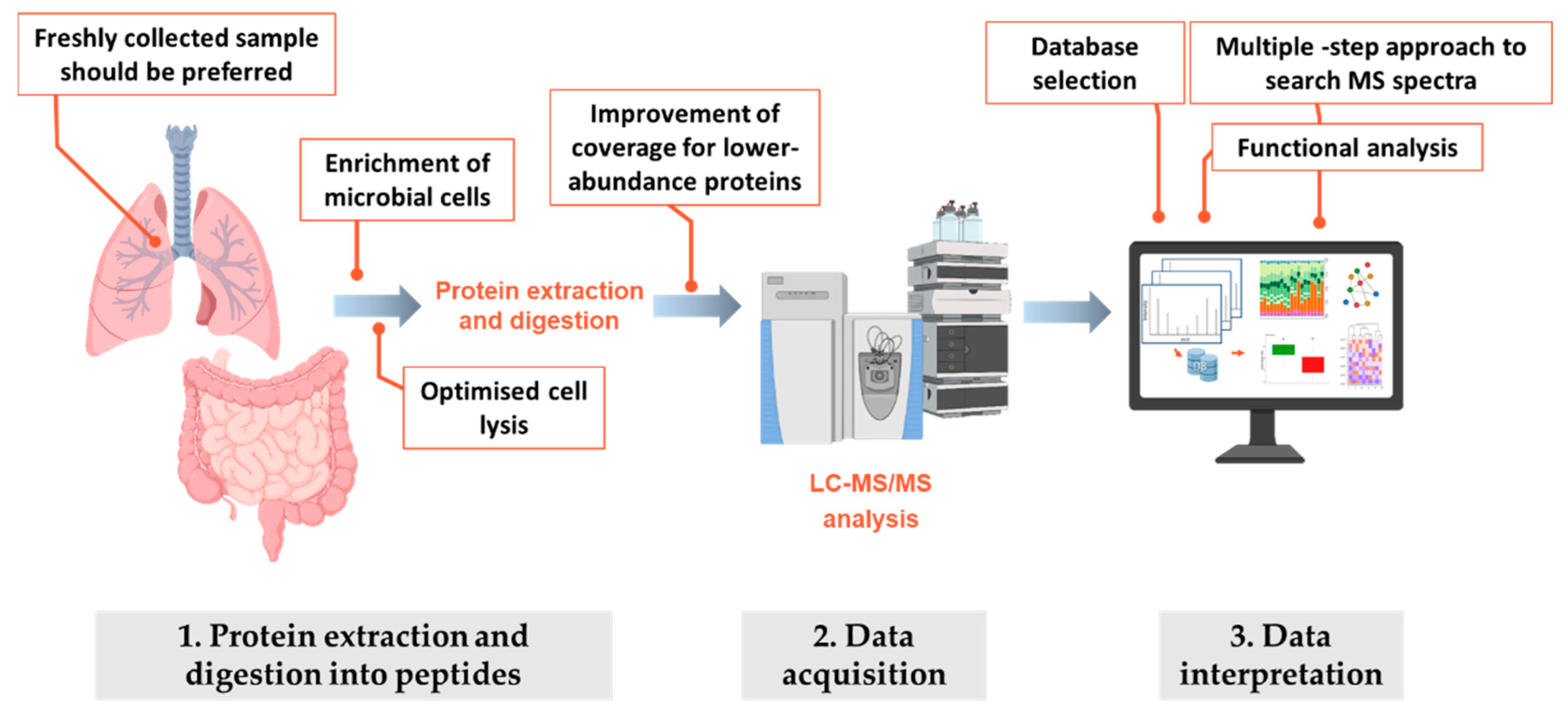
4.2. Methodological Considerations When Profiling CF Microbiota by Metaproteomics: Data Acquisition
4.3. Methodological Considerations When Profiling CF Microbiota by Metaproteomics: Data Interpretation
5. Perspectives for Applications of Proteomics-Based Approaches to CF Microbiota
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmadi, S.; Bozoky, Z.; Di Paola, M.; Xia, S.; Li, C.; Wong, A.P.; Wellhauser, L.; Molinski, S.V.; Ip, W.; Ouyang, H.; et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom. Med. 2017, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; De Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, L.; Rogers, G.B.; Walker, A.W.; Oliver, A.; Green, L.E.; Daniels, T.W.; Carroll, M.P.; Parkhill, J.; Bruce, K.D.; van der Gast, C.J. Respiratory microbiota resistance and resilience to pulmonary exacerbation and subsequent antimicrobial intervention. ISME J. 2016, 10, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, S.D.; Martiny, J.B. Colloquium paper: Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105 (Suppl. S1), 11512–11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskinen, K.; Pausan, M.R.; Perras, A.K.; Beck, M.; Bang, C.; Mora, M.; Schilhabel, A.; Schmitz, R.; Moissl-Eichinger, C. First Insights into the Diverse Human Archaeome: Specific Detection of Archaea in the Gastrointestinal Tract, Lung, and Nose and on Skin. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- van der Gast, C.J.; Walker, A.W.; Stressmann, F.A.; Rogers, G.B.; Scott, P.; Daniels, T.W.; Carroll, M.P.; Parkhill, J.; Bruce, K.D. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2011, 5, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevivino, A.; Bacci, G.; Drevinek, P.; Nelson, M.T.; Hoffman, L.; Mengoni, A. Deciphering the Ecology of Cystic Fibrosis Bacterial Communities: Towards Systems-Level Integration. Trends Mol. Med. 2019, 25, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.B.; Bruce, K.D.; Hoffman, L.R. How can the cystic fibrosis respiratory microbiome influence our clinical decision-making? Curr. Opin. Pulm. Med. 2017, 23, 536–543. [Google Scholar] [CrossRef]
- Hall, A.B.; Tolonen, A.C.; Xavier, R.J. Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet. 2017, 18, 690–699. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Héry-Arnaud, G.; Boutin, S.; Cuthbertson, L.; Elborn, S.J.; Tunney, M.M. The lung and gut microbiome: What has to be taken into consideration for cystic fibrosis? J. Cyst. Fibros. 2019, 18, 13–21. [Google Scholar] [CrossRef]
- Francoise, A.; Hery-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes 2020, 11, 536. [Google Scholar] [CrossRef]
- Lee, A.J.; Einarsson, G.G.; Gilpin, D.F.; Tunney, M.M. Multi-Omics Approaches: The Key to Improving Respiratory Health in People with Cystic Fibrosis? Front. Pharmacol. 2020, 11, 569821. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Needham, B.; Leach, S.T.; Day, A.S.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Disrupted progression of the intestinal microbiota with age in children with cystic fibrosis. Sci. Rep. 2016, 6, 24857. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, E.; Callegari, M.L.; Raia, V.; Viscovo, S.; Scotto, R.; Ferrari, S.; Morelli, L.; Buccigrossi, V.; Lo Vecchio, A.; Ruberto, E.; et al. Disrupted intestinal microbiota and intestinal inflammation in children with cystic fibrosis and its restoration with Lactobacillus GG: A randomised clinical trial. PLoS ONE 2014, 9, e87796. [Google Scholar] [CrossRef] [Green Version]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Dysbiosis of bifidobacteria and Clostridium cluster XIVa in the cystic fibrosis fecal microbiota. J. Cyst. Fibros. 2013, 12, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Duytschaever, G.; Huys, G.; Bekaert, M.; Boulanger, L.; De Boeck, K.; Vandamme, P. Cross-sectional and longitudinal comparisons of the predominant fecal microbiota compositions of a group of pediatric patients with cystic fibrosis and their healthy siblings. Appl. Environ. Microbiol. 2011, 77, 8015–8024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, S.V.; Goldfarb, K.C.; Wild, Y.K.; Kong, W.; De Lisle, R.C.; Brodie, E.L. Cystic fibrosis transmembrane conductance regulator knockout mice exhibit aberrant gastrointestinal microbiota. Gut Microbes 2013, 4, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schippa, S.; Iebba, V.; Santangelo, F.; Gagliardi, A.; De Biase, R.V.; Stamato, A.; Bertasi, S.; Lucarelli, M.; Conte, M.P.; Quattrucci, S. Cystic fibrosis transmembrane conductance regulator (CFTR) allelic variants relate to shifts in faecal microbiota of cystic fibrosis patients. PLoS ONE 2013, 8, e61176. [Google Scholar] [CrossRef]
- Hoen, A.G.; Li, J.; Moulton, L.A.; O’Toole, G.A.; Housman, M.L.; Koestler, D.C.; Guill, M.F.; Moore, J.H.; Hibberd, P.L.; Morrison, H.G.; et al. Associations between Gut Microbial Colonization in Early Life and Respiratory Outcomes in Cystic Fibrosis. J. Pediatrics 2015, 167, 138–147.e133. [Google Scholar] [CrossRef] [Green Version]
- Manor, O.; Levy, R.; Pope, C.E.; Hayden, H.S.; Brittnacher, M.J.; Carr, R.; Radey, M.C.; Hager, K.R.; Heltshe, S.L.; Ramsey, B.W.; et al. Metagenomic evidence for taxonomic dysbiosis and functional imbalance in the gastrointestinal tracts of children with cystic fibrosis. Sci. Rep. 2016, 6, 22493. [Google Scholar] [CrossRef] [Green Version]
- Ooi, C.Y.; Durie, P.R. Cystic fibrosis from the gastroenterologist’s perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 175–185. [Google Scholar] [CrossRef]
- Garg, M.; Ooi, C.Y. The Enigmatic Gut in Cystic Fibrosis: Linking Inflammation, Dysbiosis, and the Increased Risk of Malignancy. Curr. Gastroenterol. Rep. 2017, 19, 6. [Google Scholar] [CrossRef]
- Hahn, A.; Sanyal, A.; Perez, G.F.; Colberg-Poley, A.M.; Campos, J.; Rose, M.C.; Perez-Losada, M. Different next generation sequencing platforms produce different microbial profiles and diversity in cystic fibrosis sputum. J. Microbiol. Methods 2016, 130, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef]
- Lynch, J.B.; Hsiao, E.Y. Microbiomes as sources of emergent host phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H. Parkinson disease, levodopa and the gut microbiota-when microbiology meets pharmacology. Environ. Microbiol. 2020, 22, 808–812. [Google Scholar] [CrossRef]
- Cox, M.J.; Allgaier, M.; Taylor, B.; Baek, M.S.; Huang, Y.J.; Daly, R.A.; Karaoz, U.; Andersen, G.L.; Brown, R.; Fujimura, K.E.; et al. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS ONE 2010, 5, e11044. [Google Scholar] [CrossRef] [PubMed]
- Meeker, S.M.; Mears, K.S.; Sangwan, N.; Brittnacher, M.J.; Weiss, E.J.; Treuting, P.M.; Tolley, N.; Pope, C.E.; Hager, K.R.; Vo, A.T.; et al. CFTR dysregulation drives active selection of the gut microbiome. PLoS Pathog. 2020, 16, e1008251. [Google Scholar] [CrossRef]
- Dayama, G.; Priya, S.; Niccum, D.E.; Khoruts, A.; Blekhman, R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [Green Version]
- Brussow, H. The relationship between the host genome, microbiome, and host phenotype. Environ. Microbiol. 2020, 22, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieers, G.; Guery, B.; Delhaes, L. The Gut-Lung Axis in Health and Respiratory Diseases: A Place for Inter-Organ and Inter-Kingdom Crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debyser, G.; Mesuere, B.; Clement, L.; Van de Weygaert, J.; Van Hecke, P.; Duytschaever, G.; Aerts, M.; Dawyndt, P.; De Boeck, K.; Vandamme, P.; et al. Faecal proteomics: A tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debyser, G.; Aerts, M.; Van Hecke, P.; Mesuere, B.; Duytschaever, G.; Dawyndt, P.; De Boeck, K.; Vandamme, P.; Devreese, B. A Method for Comprehensive Proteomic Analysis of Human Faecal Samples to Investigate Gut Dysbiosis in Patients with Cystic Fibrosis. In Emerging Sample Treatments in Proteomics; Capelo-Martínez, J.-L., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 137–160. [Google Scholar]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic approaches in microbial ecology: An update on whole-genome and marker gene sequencing analyses. Microbial Genomics 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, P.M.; Bernard, T.; Greub, G.; Jaton, K.; Pagni, M.; Hafen, G.M. Microbiota present in cystic fibrosis lungs as revealed by whole genome sequencing. PLoS ONE 2014, 9, e90934. [Google Scholar] [CrossRef] [Green Version]
- Moran Losada, P.; Chouvarine, P.; Dorda, M.; Hedtfeld, S.; Mielke, S.; Schulz, A.; Wiehlmann, L.; Tummler, B. The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Dmitrijeva, M.; Kahlert, C.R.; Feigelman, R.; Kleiner, R.L.; Nolte, O.; Albrich, W.C.; Baty, F.; von Mering, C. Strain-Resolved Dynamics of the Lung Microbiome in Patients with Cystic Fibrosis. mBio 2021, 12. [Google Scholar] [CrossRef]
- Feigelman, R.; Kahlert, C.R.; Baty, F.; Rassouli, F.; Kleiner, R.L.; Kohler, P.; Brutsche, M.H.; von Mering, C. Sputum DNA sequencing in cystic fibrosis: Non-invasive access to the lung microbiome and to pathogen details. Microbiome 2017, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacci, G.; Taccetti, G.; Dolce, D.; Armanini, F.; Segata, N.; Di Cesare, F.; Lucidi, V.; Fiscarelli, E.; Morelli, P.; Casciaro, R.; et al. Untargeted Metagenomic Investigation of the Airway Microbiome of Cystic Fibrosis Patients with Moderate-Severe Lung Disease. Microorganisms 2020, 8, 1003. [Google Scholar] [CrossRef]
- Pust, M.-M.; Wiehlmann, L.; Davenport, C.; Rudolf, I.; Dittrich, A.-M.; Tümmler, B. The human respiratory tract microbial community structures in healthy and cystic fibrosis infants. NPJ Biofilms Microbiomes 2020, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Whelan, F.J.; Waddell, B.; Syed, S.A.; Shekarriz, S.; Rabin, H.R.; Parkins, M.D.; Surette, M.G. Culture-enriched metagenomic sequencing enables in-depth profiling of the cystic fibrosis lung microbiota. Nat. Microbiol. 2020, 5, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Shakya, M.; Lo, C.-C.; Chain, P.S.G. Advances and Challenges in Metatranscriptomic Analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [Green Version]
- Quinn, R.A.; Lim, Y.W.; Maughan, H.; Conrad, D.; Rohwer, F.; Whiteson, K.L. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. mBio 2014, 5, e00956-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobian Guemes, A.G.; Lim, Y.W.; Quinn, R.A.; Conrad, D.J.; Benler, S.; Maughan, H.; Edwards, R.; Brettin, T.; Cantu, V.A.; Cuevas, D.; et al. Cystic Fibrosis Rapid Response: Translating Multi-omics Data into Clinically Relevant Information. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteson, K.L.; Meinardi, S.; Lim, Y.W.; Schmieder, R.; Maughan, H.; Quinn, R.; Blake, D.R.; Conrad, D.; Rohwer, F. Breath gas metabolites and bacterial metagenomes from cystic fibrosis airways indicate active pH neutral 2,3-butanedione fermentation. ISME J. 2014, 8, 1247–1258. [Google Scholar] [CrossRef]
- Hahn, A.; Whiteson, K.; Davis, T.J.; Phan, J.; Sami, I.; Koumbourlis, A.C.; Freishtat, R.J.; Crandall, K.A.; Bean, H.D. Longitudinal Associations of the Cystic Fibrosis Airway Microbiome and Volatile Metabolites: A Case Study. Front. Cell. Infect. Microbiol. 2020, 10, 174. [Google Scholar] [CrossRef]
- Neerincx, A.H.; Whiteson, K.; Phan, J.L.; Brinkman, P.; Abdel-Aziz, M.I.; Weersink, E.J.M.; Altenburg, J.; Majoor, C.J.; Maitland-van der Zee, A.H.; Bos, L.D.J. Lumacaftor/ivacaftor changes the lung microbiome and metabolome in cystic fibrosis patients. J. ERJ Open Res. 2021, 7, 00731–02020. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Yang, J.; Morrissey, B.M.; Hammock, B.D.; Cross, C.E. Omics approaches in cystic fibrosis research: A focus on oxylipin profiling in airway secretions. Ann. N. Y. Acad. Sci. 2012, 1259, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Antosca, K.M.; Chernikova, D.A.; Price, C.E.; Ruoff, K.L.; Li, K.; Guill, M.F.; Sontag, N.R.; Morrison, H.G.; Hao, S.; Drumm, M.L.; et al. Altered Stool Microbiota of Infants with Cystic Fibrosis Shows a Reduction in Genera Associated with Immune Programming from Birth. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, S.; Wang, N.; Tan, H.Y.; Zhang, Z.; Feng, Y. The Cross-Talk between Gut Microbiota and Lungs in Common Lung Diseases. Front. Microbiol. 2020, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.J.; Nielsen, S.; Wemheuer, B.; Kaakoush, N.O.; Garg, M.; Needham, B.; Pickford, R.; Jaffe, A.; Thomas, T.; Ooi, C.Y. Gut Microbiota in Children with Cystic Fibrosis: A Taxonomic and Functional Dysbiosis. Sci. Rep. 2019, 9, 18593. [Google Scholar] [CrossRef] [PubMed]
- Vernocchi, P.; Del Chierico, F.; Russo, A.; Majo, F.; Rossitto, M.; Valerio, M.; Casadei, L.; La Storia, A.; De Filippis, F.; Rizzo, C.; et al. Gut microbiota signatures in cystic fibrosis: Loss of host CFTR function drives the microbiota enterophenotype. PLoS ONE 2018, 13, e0208171. [Google Scholar] [CrossRef]
- Coffey, M.J.; Garg, M.; Homaira, N.; Jaffe, A.; Ooi, C.Y. Probiotics for people with cystic fibrosis. Cochrane Database Syst. Rev. 2020, 1, CD012949. [Google Scholar] [CrossRef]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Gut Microbial-Derived Metabolomics of Asthma. Metabolites 2020, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Liessi, N.; Pedemonte, N.; Armirotti, A.; Braccia, C. Proteomics and Metabolomics for Cystic Fibrosis Research. Int. J. Mol. Sci. 2020, 21, 5439. [Google Scholar] [CrossRef] [PubMed]
- Pattison, S.H.; Gibson, D.S.; Johnston, E.; Peacock, S.; Rivera, K.; Tunney, M.M.; Pappin, D.J.; Elborn, J.S. Proteomic profile of cystic fibrosis sputum cells in adults chronically infected with Pseudomonas aeruginosa. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.; Moon, K.M.; Chen, V.; Ng, R.; Foster, L.J.; Tebbutt, S.J.; Quon, B.S. Identification of novel blood biomarkers of treatment response in cystic fibrosis pulmonary exacerbations by label-free quantitative proteomics. Sci. Rep. 2019, 9, 17126. [Google Scholar] [CrossRef] [Green Version]
- Benabdelkamel, H.; Alamri, H.; Okla, M.; Masood, A.; Abdel Jabar, M.; Alanazi, I.O.; Alfadda, A.A.; Nizami, I.; Dasouki, M.; Abdel Rahman, A.M. Serum-Based Proteomics Profiling in Adult Patients with Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 7415. [Google Scholar] [CrossRef]
- Gouveia, D.; Grenga, L.; Pible, O.; Armengaud, J. Quick microbial molecular phenotyping by differential shotgun proteomics. Environ. Microbiol. 2020, 22, 2996–3004. [Google Scholar] [CrossRef]
- Wilmes, P.; Bond, P.L. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ. Microbiol. 2004, 6, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, M. Metaproteomics: Much More than Measuring Gene Expression in Microbial Communities. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Salvato, F.; Hettich, R.L.; Kleiner, M. Five key aspects of metaproteomics as a tool to understand functional interactions in host-associated microbiomes. PLoS Pathog. 2021, 17, e1009245. [Google Scholar] [CrossRef]
- Lee, P.Y.; Chin, S.F.; Neoh, H.M.; Jamal, R. Metaproteomic analysis of human gut microbiota: Where are we heading? J. Biomed. Sci. 2017, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.D.; Viken, K.J.; Johnson, J.; McGowan, T.; Pendleton, K.M.; Griffin, T.J.; Hunter, R.C.; Rudney, J.D.; Bhargava, M. BAL Fluid Metaproteome in Acute Respiratory Failure. Am. J. Respir. Cell Mol. Biol. 2018, 59, 648–652. [Google Scholar] [CrossRef]
- Pathak, K.V.; McGilvrey, M.I.; Hu, C.K.; Garcia-Mansfield, K.; Lewandoski, K.; Eftekhari, Z.; Yuan, Y.C.; Zenhausern, F.; Menashi, E.; Pirrotte, P. Molecular Profiling of Innate Immune Response Mechanisms in Ventilator-associated Pneumonia. Mol. Cell. Proteom. 2020, 19, 1688–1705. [Google Scholar] [CrossRef]
- Lin, H.; He, Q.Y.; Shi, L.; Sleeman, M.; Baker, M.S.; Nice, E.C. Proteomics and the microbiome: Pitfalls and potential. Expert Rev. Proteom. 2019, 16, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, T.; Kunath, B.; Schallert, K.; Schäpe, S.; Abraham, P.; Armengaud, J.; Arntzen, M.; Bassignani, A.; Benndorf, D.; Fuchs, S.; et al. Critical Assessment of Metaproteome Investigation (CAMPI): A Multi-Lab Comparison of Established Workflows. bioRxiv 2021. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Budel, A.; Zoun, R.; Dorl, S.; Behne, A.; Kohrs, F.; Puttker, S.; Siewert, C.; Muth, T.; et al. A Robust and Universal Metaproteomics Workflow for Research Studies and Routine Diagnostics within 24 h Using Phenol Extraction, FASP Digest, and the MetaProteomeAnalyzer. Front. Microbiol. 2019, 10, 1883. [Google Scholar] [CrossRef] [Green Version]
- Starr, A.E.; Deeke, S.A.; Li, L.; Zhang, X.; Daoud, R.; Ryan, J.; Ning, Z.; Cheng, K.; Nguyen, L.V.H.; Abou-Samra, E.; et al. Proteomic and Metaproteomic Approaches to Understand Host-Microbe Interactions. Anal. Chem. 2018, 90, 86–109. [Google Scholar] [CrossRef]
- La Rosa, R.; Johansen, H.K.; Molin, S. Adapting to the Airways: Metabolic Requirements of Pseudomonas aeruginosa during the Infection of Cystic Fibrosis Patients. Metabolites 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribbs, S.K.; Beck, J.M. Microbiome in the pathogenesis of cystic fibrosis and lung transplant-related disease. Transl. Res. 2017, 179, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Schiebenhoefer, H.; Schallert, K.; Renard, B.Y.; Trappe, K.; Schmid, E.; Benndorf, D.; Riedel, K.; Muth, T.; Fuchs, S. A complete and flexible workflow for metaproteomics data analysis based on MetaProteomeAnalyzer and Prophane. Nat. Protoc. 2020, 15, 3212–3239. [Google Scholar] [CrossRef] [PubMed]
- Pible, O.; Allain, F.; Jouffret, V.; Culotta, K.; Miotello, G.; Armengaud, J. Estimating relative biomasses of organisms in microbiota using “phylopeptidomics”. Microbiome 2020, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Grenga, L.; Pible, O.; Armengaud, J. Pathogen proteotyping: A rapidly developing application of mass spectrometry to address clinical concerns. Clin. Mass Spectrom. 2019, 14, 9–17. [Google Scholar] [CrossRef]
- Bacci, G.; Mengoni, A.; Fiscarelli, E.; Segata, N.; Taccetti, G.; Dolce, D.; Paganin, P.; Morelli, P.; Tuccio, V.; De Alessandri, A.; et al. A Different Microbiome Gene Repertoire in the Airways of Cystic Fibrosis Patients with Severe Lung Disease. Int. J. Mol. Sci. 2017, 18, 1654. [Google Scholar] [CrossRef] [Green Version]
- Huffnagle, G.B.; Dickson, R.P.; Lukacs, N.W. The respiratory tract microbiome and lung inflammation: A two-way street. Mucosal Immunol. 2017, 10, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Molecular Level (Methodology) | Biological Questions Addressed | CF Associated Records |
|---|---|---|
| DNA (metagenomics) | What is the ecology of the CF lung microbiome and the ecological patterns of CF microbiota? | 108 |
| mRNAs (transcriptomics) | Which genes are expressed? Which components of the microbiota are active? | 3 |
| Proteins (metaproteomics) | What are the key players in the CF lung? Could such proteins be biomarkers of exacerbation? | 2 ** |
| Metabolites (metabolomics) | How does the gut microbiota affect host metabolism? | 128 |
| Lipids (lipidomics) | Is there a lipid signature associated with CF progression? | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardouin, P.; Chiron, R.; Marchandin, H.; Armengaud, J.; Grenga, L. Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives. Genes 2021, 12, 892. https://doi.org/10.3390/genes12060892
Hardouin P, Chiron R, Marchandin H, Armengaud J, Grenga L. Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives. Genes. 2021; 12(6):892. https://doi.org/10.3390/genes12060892
Chicago/Turabian StyleHardouin, Pauline, Raphael Chiron, Hélène Marchandin, Jean Armengaud, and Lucia Grenga. 2021. "Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives" Genes 12, no. 6: 892. https://doi.org/10.3390/genes12060892
APA StyleHardouin, P., Chiron, R., Marchandin, H., Armengaud, J., & Grenga, L. (2021). Metaproteomics to Decipher CF Host-Microbiota Interactions: Overview, Challenges and Future Perspectives. Genes, 12(6), 892. https://doi.org/10.3390/genes12060892