
An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story

 , and
, and
Abstract
:1. Introduction
2. Function of ABCA4
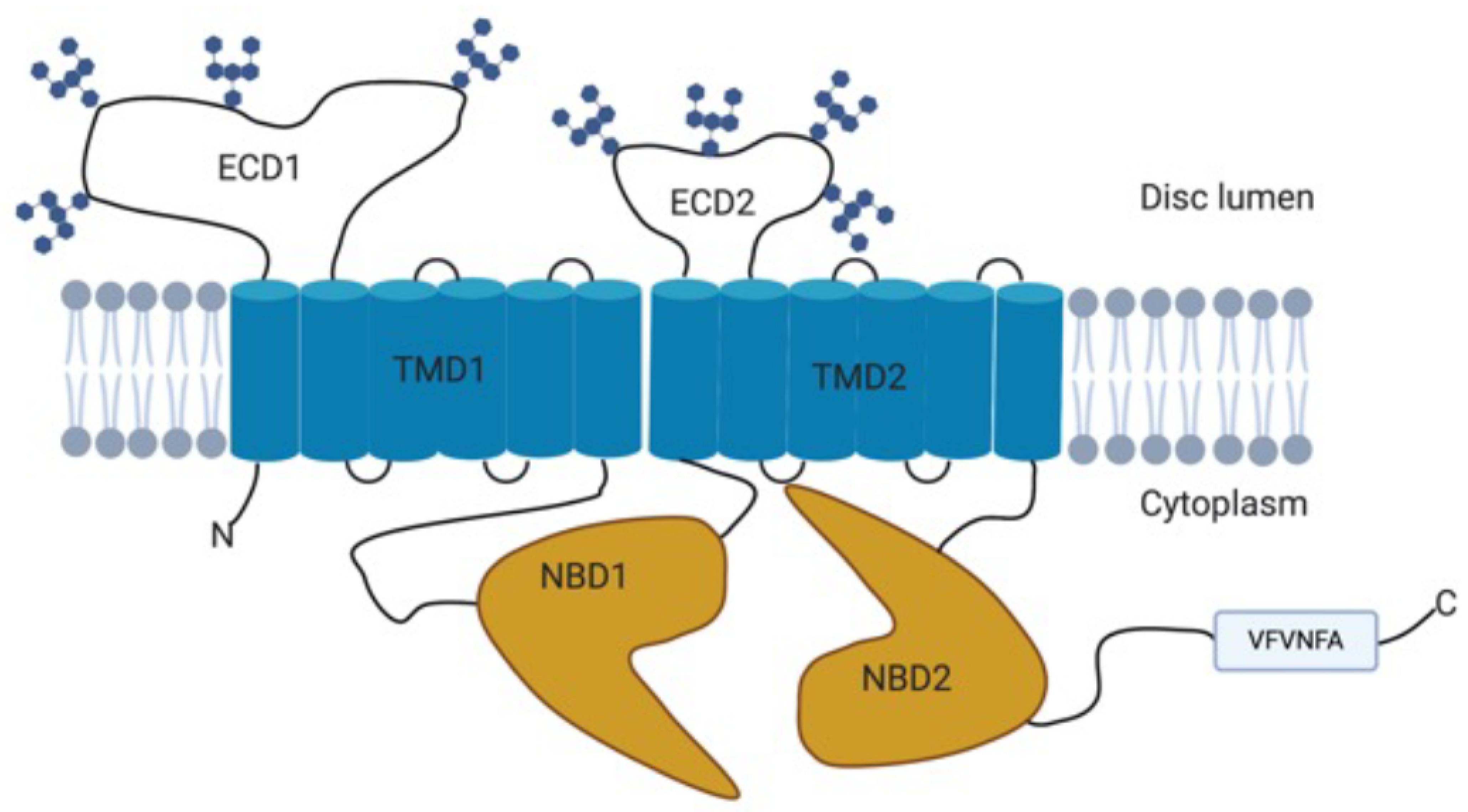
2.1. Structure
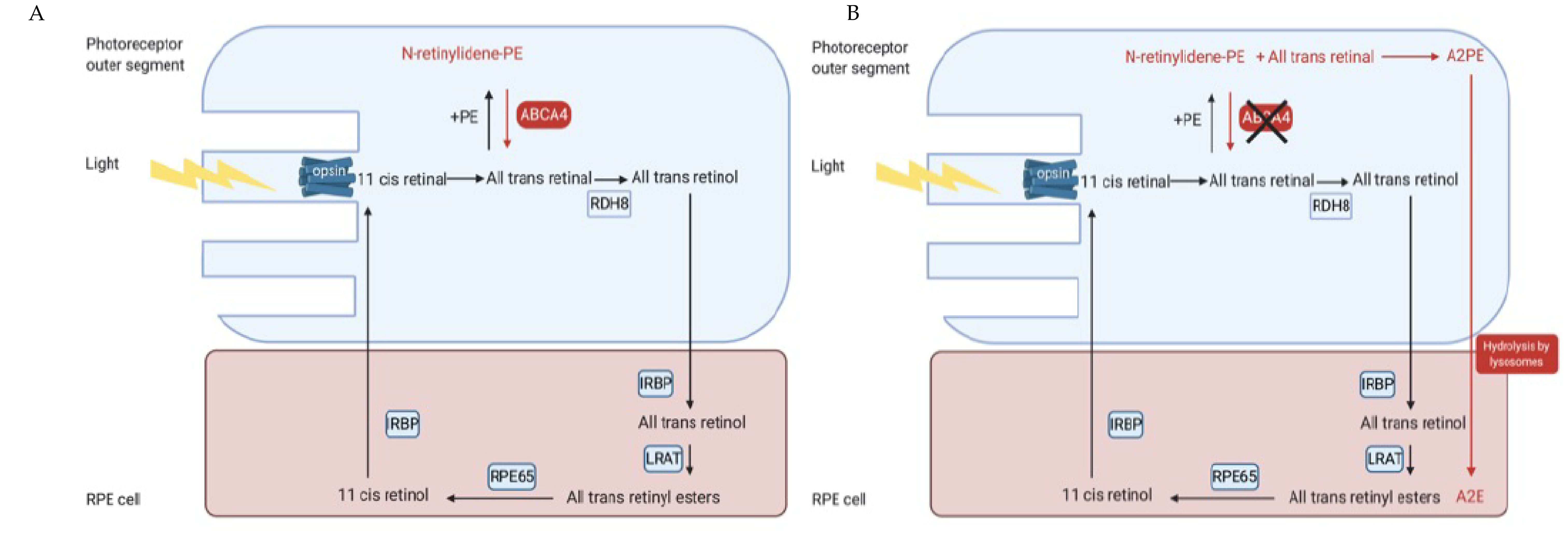
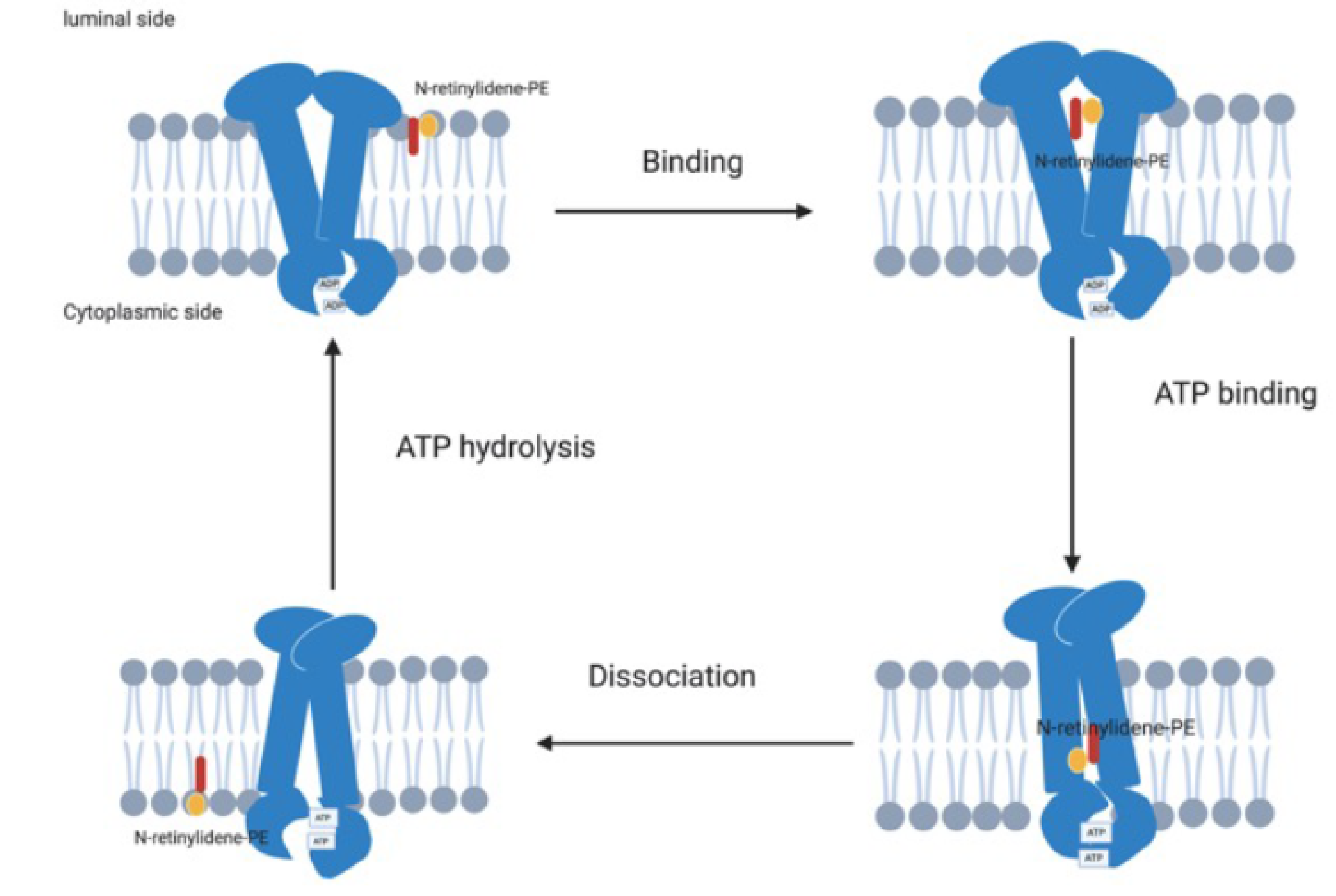
2.2. Function and Role within the Visual Cycle
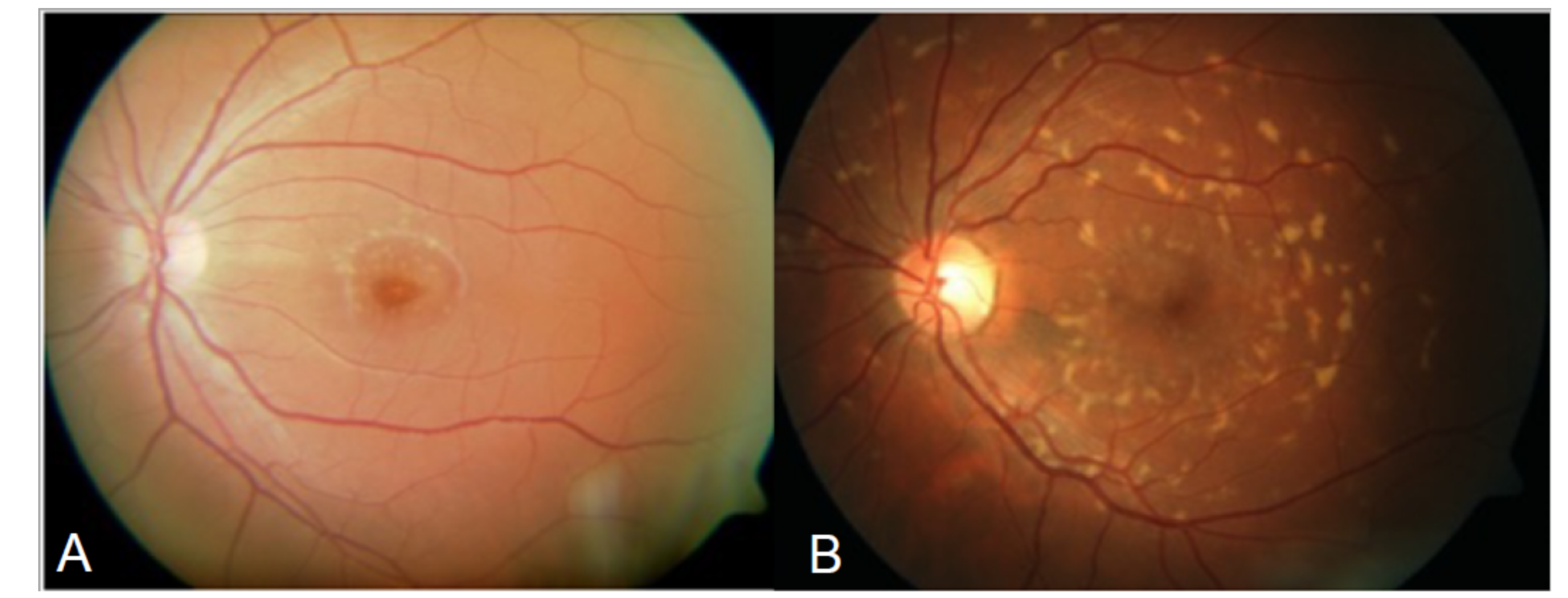
3. Clinical Phenotypes and Phenocopies in ABCA4 Retinopathies (ABCA4R)
4. Genetics of ABCA4R
4.1. Genetic Testing in ABCA4
4.2. Spectrum of Pathogenic Variants in ABCA4
4.3. Variability in Variants between Populations
4.4. Missing Heritability
4.4.1. Deep Intronic Variants
4.4.2. Structural Variants
4.4.3. Hypomorphic Alleles and Modifiers
5. Pathogenicity and Severity of ABCA4 Variants
5.1. Predicting Pathogenicity
5.2. Functional Analysis of ABCA4 Variants
6. Therapies
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blacharski, P. Retinal Dystrophies and Degenerations; Raven Press: New York, NY, USA, 1988; pp. 135–159. [Google Scholar]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Yatsenko, A.N.; Shroyer, N.F.; Lewis, R.A.; Lupski, J.R. Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum. Genet. 2001, 108, 346–355. [Google Scholar] [CrossRef]
- Jaakson, K.; Zernant, J.; Külm, M.; Hutchinson, A.; Tonisson, N.; Glavac, D.; Ravnik-Glavac, M.; Hawlina, M.; Meltzer, M.R.; Caruso, R.C.; et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 2003, 22, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Stargardt, K. Über familiäre, progressive Degeneration in der Maculagegend des Auges. Albrecht Graefes Arch. Ophthalmol. 1909, 71, 534–550. [Google Scholar] [CrossRef] [Green Version]
- Franceschetti, A.; Francois, J. Fundus flavimaculatus. Arch. Ophtalmol. Rev. Gen. Ophtalmol. 1965, 25, 505. [Google Scholar] [PubMed]
- Franceschetti, A. A special form of tapetoretinal degeneration: Fundus flavimaculatus. Trans. Am. Acad. Ophthalmol. Otolaryngol. 1965, 69, 1048–1053. [Google Scholar]
- Kaplan, J.; Gerber, S.; Larget-Piet, D.; Rozet, J.-M.; Dollfus, H.; Dufier, J.-L.; Odent, S.; Postel-Vinay, A.; Janin, N.; Briard, M.-L.; et al. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat. Genet. 1993, 5, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Hadden, O.B.; Gass, J.D. Fundus flavimaculatus and Stargardt’s disease. Am. J. Ophthalmol. 1976, 82, 527–539. [Google Scholar] [CrossRef]
- Noble, K.G.; Carr, R.E. Stargardt’s disease and fundus flavimaculatus. Arch. Ophthalmol. 1979, 97, 1281–1285. [Google Scholar] [CrossRef]
- Gerber, S.; Rozet, J.M.; Bonneau, D.; Souied, E.; Camuzat, A.; Dufier, J.L.; Amalric, P.; Weissenbach, J.; Munnich, A.; Kaplan, J. A gene for late-onset fundus flavimaculatus with macular dystrophy maps to chromosome 1p13. Am. J. Hum. Genet. 1995, 56, 396–399. [Google Scholar]
- Hoyng, C.B.; Poppelaars, F.; Van de Pol, T.J.R.; Kremer, H.; Pinckers, A.J.L.G.; Deutman, A.F.; Cremers, F.P.M. Genetic fine mapping of the gene for recessive Stargardt disease. Hum. Genet. 1996, 98, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa rim protein of retinal rod outer segments is a member of the ABC transporter superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Nathans, J. Stargardt’s ABCR is localized to the disc membrane of retinal rod outer segments. Nat. Genet. 1997, 17, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Molday, L.L.; Rabin, A.R.; Molday, R.S. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet. 2000, 25, 257–258. [Google Scholar] [CrossRef]
- Lois, N.; Halfyard, A.S.; Bird, A.C.; Holder, G.E.; Fitzke, F.W. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am. J. Ophthalmol. 2004, 138, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [Green Version]
- Schwoerer, J.; Secretan, M.; Zografos, L.; Piguet, B. Indocyanine green angiography in Fundus flavimaculatus. Ophthalmologica 2000, 214, 240–245. [Google Scholar] [CrossRef]
- Khan, K.N.; Kasilian, M.; Mahroo, O.A.R.; Tanna, P.; Kalitzeos, A.; Robson, A.G.; Tsunoda, K.; Iwata, T.; Moore, A.T.; Fujinami, K.; et al. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology 2018, 125, 735–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, K.; Seong, M.-W.; Park, K.H.; Park, S.S.; Woo, S.J. Genotypic profile and phenotype correlations of ABCA4-associated retinopathy in Koreans. Mol. Vis. 2019, 25, 679–690. [Google Scholar]
- Al-Ani, H.H.; Sheck, L.; Vincent, A.L. Peripheral pigmented lesions in ABCA4-associated retinopathy. Ophthalmic Genet. 2021, 42, 383–391. [Google Scholar] [CrossRef]
- Kong, X.; Strauss, R.W.; Michaelides, M.; Cideciyan, A.V.; Sahel, J.-A.; Muñoz, B.; West, S.; Scholl, H.P.N.; Wolfson, Y.; Bittencourt, M.; et al. Visual Acuity Loss and Associated Risk Factors in the Retrospective Progression of Stargardt Disease Study (ProgStar Report No. 2). Ophthalmology 2016, 123, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Strauss, R.W.; Cideciyan, A.V.; Michaelides, M.; Sahel, J.-A.; Munoz, B.; Ahmed, M.; Ervin, A.M.; West, S.K.; Cheetham, J.K.; et al. Visual Acuity Change over 12 Months in the Prospective Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: ProgStar Report Number 6. Ophthalmology 2017, 124, 1640–1651. [Google Scholar] [CrossRef] [Green Version]
- Lambertus, S.; Van Huet, R.A.; Bax, N.M.; Hoefsloot, L.H.; Cremers, F.P.; Boon, C.J.; Klevering, B.J.; Hoyng, C.B. Early-onset stargardt disease: Phenotypic and genotypic characteristics. Ophthalmology 2015, 122, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Utz, V.M.; Coussa, R.G.; Marino, M.J.; Chappelow, A.V.; Pauer, G.J.; Hagstrom, S.A.; Traboulsi, E.I. Predictors of visual acuity and genotype-phenotype correlates in a cohort of patients with Stargardt disease. Br. J. Ophthalmol. 2014, 98, 513–518. [Google Scholar] [CrossRef]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501. [Google Scholar] [CrossRef]
- Haaften, S.C.W.-V.; Boon, C.J.; Cremers, F.P.; Hoefsloot, L.H.; Den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; West, S.K.; Strauss, R.W.; Munoz, B.; Cideciyan, A.V.; Michaelides, M.; Ho, A.; Ahmed, M.; Schönbach, E.M.; Cheetham, J.K.; et al. Progression of Visual Acuity and Fundus Autofluorescence in Recent-Onset Stargardt Disease: ProgStar Study Report #4. Ophthalmol. Retina 2017, 1, 514–523. [Google Scholar] [CrossRef]
- Testa, F.; Melillo, P.; Di Iorio, V.; Orrico, A.; Attanasio, M.; Rossi, S.; Simonelli, F. Macular function and morphologic features in juvenile stargardt disease: Longitudinal study. Ophthalmology 2014, 121, 2399–2405. [Google Scholar] [CrossRef] [Green Version]
- Al-khuzaei, S.; Shah, M.; Foster, C.R.; Yu, J.; Broadgate, S.; Halford, S.; Downes, S.M. The role of multimodal imaging and vision function testing in ABCA4 related retinopathies and their relevance to future therapeutic interventions. Ther. Adv. Ophthalmol. 2021. In press. [Google Scholar]
- Stone, E.M.; Nichols, B.E.; Kimura, A.E.; Weingeist, T.A.; Drack, A.; Sheffield, V.C. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch. Ophthalmol. 1994, 112, 765–772. [Google Scholar] [CrossRef]
- Kniazeva, M.; Chiang, M.F.; Morgan, B.; Anduze, A.L.; Zack, D.J.; Han, M.; Zhang, K. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am. J. Hum. Genet. 1999, 64, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsybovsky, Y.; Molday, R.S.; Palczewski, K. The ATP-binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol. 2010, 703, 105–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linton, K.J. Structure and function of ABC transporters. Physiology 2007, 22, 122–130. [Google Scholar] [CrossRef]
- Molday, R.S.; Zhang, K. Defective lipid transport and biosynthesis in recessive and dominant Stargardt macular degeneration. Prog. Lipid Res. 2010, 49, 476–492. [Google Scholar] [CrossRef] [Green Version]
- Tsybovsky, Y.; Orban, T.; Molday, R.S.; Taylor, D.; Palczewski, K. Molecular Organization and ATP-Induced Conformational Changes of ABCA4, the Photoreceptor-Specific ABC Transporter. Structure 2013, 21, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Molday, R.S. Chapter Twenty-Four—Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. In Progress in Molecular Biology and Translational Science; Hejtmancik, J.F., Nickerson, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2015; Volume 134, pp. 415–431. [Google Scholar]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattner, A.; Smallwood, P.M.; Nathans, J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J. Biol. Chem. 2000, 275, 11034–11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBee, J.K.; Palczewski, K.; Baehr, W.; Pepperberg, D.R. Confronting complexity: The interlink of phototransduction and retinoid metabolism in the vertebrate retina. Prog. Retin. Eye Res. 2001, 20, 469–529. [Google Scholar] [CrossRef]
- Saari, J.C. Biochemistry of visual pigment regeneration: The Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 2000, 41, 337–348. [Google Scholar]
- Poincelot, R.P.; Millar, P.G.; Kimbel, R.L., Jr.; Abrahamson, E.W. Lipid to protein chromophore transfer in the photolysis of visual pigments. Nature 1969, 221, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.E.; Maude, M.B. Phospholipids of bovine outer segments. Biochemistry 1970, 9, 3624–3628. [Google Scholar] [CrossRef]
- Beharry, S.; Zhong, M.; Molday, R.S. N-retinylidene-phosphatidylethanolamine is the preferred retinoid substrate for the photoreceptor-specific ABC transporter ABCA4 (ABCR). J. Biol. Chem. 2004, 279, 53972–53979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef] [Green Version]
- Pollock, N.L.; Callaghan, R. The lipid translocase, ABCA4: Seeing is believing. FEBS J. 2011, 278, 3204–3214. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Parish, C.A.; Vollmer, H.R.; Itagaki, Y.; Fishkin, N.; Nakanishi, K.; Sparrow, J.R. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J. Biol. Chem. 2002, 277, 7183–7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, J.R.; Kim, S.R.; Cuervo, A.M.; Bandhyopadhyayand, U. A2E, a pigment of RPE lipofuscin, is generated from the precursor, A2PE by a lysosomal enzyme activity. Adv. Exp. Med. Biol. 2008, 613, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 7154–7159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Eldred, G.E.; Lasky, M.R. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature 1993, 361, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Quazi, F.; Molday, R.S. ATP-binding cassette transporter ABCA4 and chemical isomerization protect photoreceptor cells from the toxic accumulation of excess 11-cis-retinal. Proc. Natl. Acad. Sci. USA 2014, 111, 5024–5029. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, A.; Van Driel, M.A.; Van de Pol, D.J.; Klevering, B.J.; Van Haren, F.J.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; Pinckers, A.J.; et al. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 1999, 64, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cremers, F.P.M. ABCA4-Associated Stargardt Disease. Klin. Monbl. Augenheilkd. 2020, 237, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Sears, A.E.; Bernstein, P.S.; Cideciyan, A.V.; Hoyng, C.; Issa, P.C.; Palczewski, K.; Rosenfeld, P.J.; Sadda, S.; Schraermeyer, U.; Sparrow, J.R.; et al. Towards Treatment of Stargardt Disease: Workshop Organized and Sponsored by the Foundation Fighting Blindness. Transl. Vis. Sci. Technol. 2017, 6, 6. [Google Scholar] [CrossRef]
- Duncker, T.; Tsang, S.H.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative Fundus Autofluorescence Distinguishes ABCA4-Associated and Non–ABCA4-Associated Bull’s-Eye Maculopathy. Ophthalmology 2015, 122, 345–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noupuu, K.; Lee, W.; Zernant, J.; Greenstein, V.C.; Tsang, S.; Allikmets, R. Recessive Stargardt disease phenocopying hydroxychloroquine retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2016, 254, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; Del Rio, T.; Dean, M.; Vilageliu, L.; Gonzàlez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet. 1998, 18, 11–12. [Google Scholar] [CrossRef]
- Fukui, T.; Yamamoto, S.; Nakano, K.; Tsujikawa, M.; Morimura, H.; Nishida, K.; Ohguro, N.; Fujikado, T.; Irifune, M.; Kuniyoshi, K.; et al. ABCA4 gene mutations in Japanese patients with Stargardt disease and retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2819–2824. [Google Scholar]
- Simonelli, F.; Testa, F.; Zernant, J.; Nesti, A.; Rossi, S.; Rinaldi, E.; Allikmets, R. Association of a homozygous nonsense mutation in the ABCA4 (ABCR) gene with cone-rod dystrophy phenotype in an Italian family. Ophthalmic Res. 2004, 36, 82–88. [Google Scholar] [CrossRef]
- Lee, W.; Zernant, J.; Nagasaki, T.; Tsang, S.H.; Allikmets, R. Deep Scleral Exposure: A Degenerative Outcome of End-Stage Stargardt Disease. Am. J. Ophthalmol. 2018, 195, 16–25. [Google Scholar] [CrossRef]
- Tanaka, K.; Lee, W.; Zernant, J.; Schuerch, K.; Ciccone, L.; Tsang, S.H.; Sparrow, J.R.; Allikmets, R. The Rapid-Onset Chorioretinopathy Phenotype of ABCA4 Disease. Ophthalmology 2018, 125, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Kniazeva, M.; Han, M.; Li, W.; Yu, Z.; Yang, Z.; Li, Y.; Metzker, M.L.; Allikmets, R.; Zack, D.J.; et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 2001, 27, 89–93. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Investig. 2008, 118, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Bither, P.P.; Park, R.; Donoso, L.A.; Seidman, J.G.; Seidman, C.E. A dominant Stargardt’s macular dystrophy locus maps to chromosome 13q34. Arch. Ophthalmol. 1994, 112, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, A.; Meire, F.; Hoyng, C.B.; Vink, C.; Van Regemorter, N.; Karan, G.; Yang, Z.; Cremers, F.P.M.; Zhang, K. A Novel Mutation in the ELOVL4 Gene Causes Autosomal Dominant Stargardt-like Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4263–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Pozo-Valero, M.; Martin-Merida, I.; Jimenez-Rolando, B.; Arteche, A.; Avila-Fernandez, A.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Van Cauwenbergh, C.; De Baere, E.; Rivolta, C.; et al. Expanded Phenotypic Spectrum of Retinopathies Associated with Autosomal Recessive and Dominant Mutations in PROM1. Am. J. Ophthalmol. 2019, 207, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Johnson, S.; Poulson, A.; Bradshaw, K.; Bellmann, C.; Hunt, D.M.; Moore, A.T. An autosomal dominant bull’s-eye macular dystrophy (MCDR2) that maps to the short arm of chromosome 4. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1657–1662. [Google Scholar] [CrossRef] [Green Version]
- Boon, C.J.; Van Schooneveld, M.J.; Den Hollander, A.I.; Van Lith-Verhoeven, J.J.; Zonneveld-Vrieling, M.N.; Theelen, T.; Cremers, F.P.; Hoyng, C.B.; Klevering, B.J. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br. J. Ophthalmol. 2007, 91, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibanez Iv, M.B.; De Guimarães, T.A.C.; Capasso, J.; Bello, N.; Levin, A.V. Stargardt misdiagnosis: How ocular genetics helps. Am. J. Med. Genet. Part A 2020, 185, 814–819. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Hudspith, K.A.Z.; Broadgate, S.; Shanks, M.E.; Clouston, P.; Németh, A.H.; Halford, S.; Downes, S.M. Targeted next generation sequencing and family survey enable correct genetic diagnosis in CRX associated macular dystrophy—A case report. BMC Ophthalmol. 2021, 21, 168. [Google Scholar] [CrossRef] [PubMed]
- Wolock, C.J.; Stong, N.; Ma, C.J.; Nagasaki, T.; Lee, W.; Tsang, S.H.; Kamalakaran, S.; Goldstein, D.B.; Allikmets, R. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet. Med. 2019, 21, 2336–2344. [Google Scholar] [CrossRef]
- Shah, M.; Broadgate, S.; Shanks, M.; Clouston, P.; Yu, J.; MacLaren, R.E.; Németh, A.H.; Halford, S.; Downes, S.M. Association of Clinical and Genetic Heterogeneity With BEST1 Sequence Variations. JAMA Ophthalmol. 2020, 138, 544–551. [Google Scholar] [CrossRef]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2019, 104, 451–460. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Robson, A.G.; Broadgate, S.; Plagnol, V.; McKibbin, M.; Halford, S.; Michaelides, M.; Holder, G.E.; Moore, A.T.; et al. Characterization of CDH3-Related Congenital Hypotrichosis With Juvenile Macular Dystrophy. JAMA Ophthalmol. 2016, 134, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halford, S.; Holt, R.; Németh, A.H.; Downes, S.M. Homozygous Deletion in CDH3 and Hypotrichosis With Juvenile Macular Dystrophy. Arch. Ophthalmol. 2012, 130, 1490–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncker, T.; Tsang, S.H.; Woods, R.L.; Lee, W.; Zernant, J.; Allikmets, R.; Delori, F.C.; Sparrow, J.R. Quantitative Fundus Autofluorescence and Optical Coherence Tomography in PRPH2/RDS- and ABCA4-Associated Disease Exhibiting Phenotypic Overlap. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3159–3170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, I.H.; Sharma, S.; Luqmani, R.; Downes, S.M. Hydroxychloroquine retinopathy. Eye 2017, 31, 828–845. [Google Scholar] [CrossRef]
- Mittra, R.; Mieler, W. Drug Toxicity of the Posterior Segment. In Retina, 5th ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; Volume 2, pp. 1532–1554. [Google Scholar] [CrossRef]
- Tracewska, A.M.; Kocyła-Karczmarewicz, B.; Rafalska, A.; Murawska, J.; Jakubaszko-Jablonska, J.; Rydzanicz, M.; Stawiński, P.; Ciara, E.; Khan, M.I.; Henkes, A.; et al. Genetic Spectrum of ABCA4-Associated Retinal Degeneration in Poland. Genes 2019, 10, 959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodi, A.; Bini, A.; Passerini, I.; Puccioni, M.; Torricelli, F.; Menchini, U. Variable expressivity of abca4 gene mutations in an italian family with stargardt disease. Retin. Cases Brief Rep. 2008, 2, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, D.; Runhart, E.H.; Bax, N.M.; Liefers, B.; Lambertus, S.L.; Sanchez, C.I.; Cremers, F.P.M.; Hoyng, C.B. Highly Variable Disease Courses in Siblings with Stargardt Disease. Ophthalmology 2019, 126, 1712–1721. [Google Scholar] [CrossRef] [Green Version]
- Passerini, I.; Sodi, A.; Giambene, B.; Mariottini, A.; Menchini, U.; Torricelli, F. Novel mutations in of the ABCR gene in italian patients with Stargardt disease. Eye 2010, 24, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runhart, E.H.; Sangermano, R.; Cornelis, S.S.; Verheij, J.B.G.M.; Plomp, A.S.; Boon, C.J.F.; Lugtenberg, D.; Roosing, S.; Bax, N.M.; Blokland, E.A.W.; et al. The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans With Severe Variants. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3220–3231. [Google Scholar] [CrossRef] [Green Version]
- Runhart, E.H.; Khan, M.; Cornelis, S.S.; Roosing, S.; Del Pozo-Valero, M.; Lamey, T.M.; Liskova, P.; Roberts, L.; Stöhr, H.; Klaver, C.C.W.; et al. Association of Sex With Frequent and Mild ABCA4 Alleles in Stargardt Disease. JAMA Ophthalmol. 2020, 138, 1035–1042. [Google Scholar] [CrossRef]
- Lee, W.; Zernant, J.; Nagasaki, T.; Allikmets, R. Reevaluating the Association of Sex With ABCA4 Alleles in Patients With Stargardt Disease. JAMA Ophthalmol. 2021, 139, 654. [Google Scholar] [CrossRef]
- Gerber, S.; Rozet, J.M.; Van de Pol, T.J.; Hoyng, C.B.; Munnich, A.; Blankenagel, A.; Kaplan, J.; Cremers, F.P. Complete exon-intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease. Genomics 1998, 48, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Ernest, P.J.G.; Boon, C.J.F.; Klevering, B.J.; Hoefsloot, L.H.; Hoyng, C.B. Outcome of ABCA4 microarray screening in routine clinical practice. Mol. Vis. 2009, 15, 2841–2847. [Google Scholar] [PubMed]
- Aguirre-Lamban, J.; Riveiro-Alvarez, R.; Maia-Lopes, S.; Cantalapiedra, D.; Vallespin, E.; Avila-Fernandez, A.; Villaverde-Montero, C.; Trujillo-Tiebas, M.J.; Ramos, C.; Ayuso, C. Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: Identification of 21 novel variants. Br. J. Ophthalmol. 2009, 93, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Duno, M.; Schwartz, M.; Larsen, P.L.; Rosenberg, T. Phenotypic and genetic spectrum of Danish patients with ABCA4-related retinopathy. Ophthalmic Genet. 2012, 33, 225–231. [Google Scholar] [CrossRef]
- Shroyer, N.F.; Lewis, R.A.; Yatsenko, A.N.; Wensel, T.G.; Lupski, J.R. Cosegregation and functional analysis of mutant ABCR (ABCA4) alleles in families that manifest both Stargardt disease and age-related macular degeneration. Hum. Mol. Genet. 2001, 10, 2671–2678. [Google Scholar] [CrossRef] [Green Version]
- Strom, S.P.; Gao, Y.-Q.; Martinez, A.; Ortube, C.; Chen, Z.; Nelson, S.F.; Nusinowitz, S.; Farber, D.B.; Gorin, M.B. Molecular diagnosis of putative Stargardt disease probands by exome sequencing. BMC Med. Genet. 2012, 13, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.; Choi, S.W.; Shim, S.H.; Song, W.K. Clinical and Genetic Characteristics Analysis of Korean Patients with Stargardt Disease Using Targeted Exome Sequencing. Ophthalmologica 2019, 241, 38–48. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; Van de Vorst, M.; Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.D.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Valverde, D.; Riveiro-Alvarez, R.; Bernal, S.; Jaakson, K.; Baiget, M.; Navarro, R.; Ayuso, C. Microarray-based mutation analysis of the ABCA4 gene in Spanish patients with Stargardt disease: Evidence of a prevalent mutated allele. Mol. Vis. 2006, 12, 902–908. [Google Scholar] [PubMed]
- Rosenberg, T.; Klie, F.; Garred, P.; Schwartz, M. N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis. 2007, 13, 1962–1969. [Google Scholar] [PubMed]
- Maia-Lopes, S.; Aguirre-Lamban, J.; Castelo-Branco, M.; Riveiro-Alvarez, R.; Ayuso, C.; Silva, E.D. ABCA4 mutations in Portuguese Stargardt patients: Identification of new mutations and their phenotypic analysis. Mol. Vis. 2009, 15, 584–591. [Google Scholar] [PubMed]
- Bertelsen, M.; Zernant, J.; Larsen, M.; Duno, M.; Allikmets, R.; Rosenberg, T. Generalized choriocapillaris dystrophy, a distinct phenotype in the spectrum of ABCA4-associated retinopathies. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2766–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort—Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.-Y.; Li, J.-K.; Gao, F.-J.; Qi, Y.-H.; Xu, P.; Zhang, Y.-J.; Wang, D.-D.; Wang, L.-S.; Li, W.; Wang, M.; et al. ABCA4 Gene Screening in a Chinese Cohort With Stargardt Disease: Identification of 37 Novel Variants. Front. Genet. 2019, 10, 773. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Strauss, R.W.; Chiang, J.P.; Audo, I.S.; Bernstein, P.S.; Birch, D.G.; Bomotti, S.M.; Cideciyan, A.V.; Ervin, A.M.; Marino, M.J.; et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 2019, 103, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, F.; Westin, I.M.; Österman, L.; Sandgren, O.; Burstedt, M.; Holmberg, M.; Golovleva, I. ATP-binding cassette subfamily A, member 4 intronic variants c.4773+3A>G and c.5461-10T>C cause Stargardt disease due to defective splicing. Acta Ophthalmol. 2018, 96, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; Van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.; Goercharn-Ramlal, A.S.; Dijk, A.H.D.E.-V.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Schindler, E.I.; Nylen, E.L.; Ko, A.C.; Affatigato, L.M.; Heggen, A.C.; Wang, K.; Sheffield, V.C.; Stone, E.M. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum. Mol. Genet. 2010, 19, 3693–3701. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Mackay, D.S.; Tsunoda, K.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Moore, A.T.; Michaelides, M.; et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 2013, 120, 2324–2331. [Google Scholar] [CrossRef]
- Escaramís, G.; Docampo, E.; Rabionet, R. A decade of structural variants: Description, history and methods to detect structural variation. Brief. Funct. Genom. 2015, 14, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Yatsenko, A.N.; Shroyer, N.F.; Lewis, R.A.; Lupski, J.R. An ABCA4 genomic deletion in patients with Stargardt disease. Hum. Mutat. 2003, 21, 636–644. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Zernant, J.; Collison, F.T.; Lee, W.; Fishman, G.A.; Noupuu, K.; Yuan, B.; Cai, C.; Lupski, J.R.; Yannuzzi, L.A.; Tsang, S.H.; et al. Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum. Mutat. 2014, 35, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.J.; Nossek, C.A.; Greenberg, L.J.; Ramesar, R.S. Stargardt macular dystrophy: Common ABCA4 mutations in South Africa--establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 2012, 18, 280–289. [Google Scholar]
- Heathfield, L.; Lacerda, M.; Nossek, C.; Roberts, L.; Ramesar, R.S. Stargardt disease: Towards developing a model to predict phenotype. Eur. J. Hum. Genet. 2013, 21, 1173–1176. [Google Scholar] [CrossRef]
- Green, J.S.; O’Rielly, D.D.; Pater, J.A.; Houston, J.; Rajabi, H.; Galutira, D.; Benteau, T.; Sheaves, A.; Abdelfatah, N.; Bautista, D.; et al. The genetic architecture of Stargardt macular dystrophy (STGD1): A longitudinal 40-year study in a genetic isolate. Eur J. Hum. Genet. 2020, 28, 925–937. [Google Scholar] [CrossRef]
- Zaneveld, J.; Siddiqui, S.; Li, H.; Wang, X.; Wang, H.; Wang, K.; Li, H.; Ren, H.; Lopez, I.; Dorfman, A.; et al. Comprehensive analysis of patients with Stargardt macular dystrophy reveals new genotype–phenotype correlations and unexpected diagnostic revisions. Genet. Med. 2015, 17, 262–270. [Google Scholar] [CrossRef] [Green Version]
- Ścieżyńska, A.; Oziębło, D.; Ambroziak, A.M.; Korwin, M.; Szulborski, K.; Krawczyński, M.; Stawiński, P.; Szaflik, J.; Szaflik, J.P.; Płoski, R.; et al. Next-generation sequencing of ABCA4: High frequency of complex alleles and novel mutations in patients with retinal dystrophies from Central Europe. Exp. Eye Res. 2016, 145, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; White, K.; Stohr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacón-Camacho, O.F.; Granillo-Alvarez, M.; Ayala-Ramírez, R.; Zenteno, J.C. ABCA4 mutational spectrum in Mexican patients with Stargardt disease: Identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp. Eye Res. 2013, 109, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Shroyer, N.F.; Singh, N.; Allikmets, R.; Hutchinson, A.; Li, Y.; Lupski, J.R.; Leppert, M.; Dean, M. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 1999, 64, 422–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, C.E.; Rucinski, D.; Rosenfeld, P.J.; Hirose, T.; Berson, E.L.; Dryja, T.P. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2229–2236. [Google Scholar]
- Hargitai, J.; Zernant, J.; Somfai, G.M.; Vamos, R.; Farkas, A.; Salacz, G.; Allikmets, R. Correlation of clinical and genetic findings in Hungarian patients with Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4402–4408. [Google Scholar] [CrossRef] [Green Version]
- September, A.V.; Vorster, A.A.; Ramesar, R.S.; Greenberg, L.J. Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Downes, S.M.; Packham, E.; Cranston, T.; Clouston, P.; Seller, A.; Németh, A.H. Detection rate of pathogenic mutations in ABCA4 using direct sequencing: Clinical and research implications. Arch. Ophthalmol. 2012, 130, 1486–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassisi, M.; Mohand-Saïd, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Méjécase, C.; Varin, J.; Wohlschlegel, J.; Dhaenens, C.-M.; Sahel, J.-A.; et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. Int. J. Mol. Sci. 2019, 20, 5053. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef]
- Fingert, J.H.; Eliason, D.A.; Phillips, N.C.; Lotery, A.J.; Sheffield, V.C.; Stone, E.M. Case of Stargardt disease caused by uniparental isodisomy. Arch. Ophthalmol. 2006, 124, 744–745. [Google Scholar] [CrossRef] [Green Version]
- Riveiro-Alvarez, R.; Valverde, D.; Lorda-Sanchez, I.; Trujillo-Tiebas, M.J.; Cantalapiedra, D.; Vallespin, E.; Aguirre-Lamban, J.; Ramos, C.; Ayuso, C. Partial paternal uniparental disomy (UPD) of chromosome 1 in a patient with Stargardt disease. Mol. Vis. 2007, 13, 96–101. [Google Scholar] [PubMed]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; Van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002733. [Google Scholar] [CrossRef] [Green Version]
- Runhart, E.H.; Valkenburg, D.; Cornelis, S.S.; Khan, M.; Sangermano, R.; Albert, S.; Bax, N.M.; Astuti, G.D.N.; Gilissen, C.; Pott, J.-W.R.; et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4249–4256. [Google Scholar] [CrossRef] [Green Version]
- Holtan, J.P.; Aukrust, I.; Jansson, R.W.; Berland, S.; Bruland, O.; Gjerde, B.L.; Stokowy, T.; Bojovic, O.; Forsaa, V.; Austeng, D.; et al. Clinical features and molecular genetics of patients with ABCA4-retinal dystrophies. Acta Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Bauwens, M.; De Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; De Jaegere, S.; De Ravel, T.; De Rademaeker, M.; et al. An Augmented ABCA4 Screen Targeting Noncoding Regions Reveals a Deep Intronic Founder Variant in Belgian Stargardt Patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef]
- Stenirri, S.; Battistella, S.; Fermo, I.; Manitto, M.P.; Martina, E.; Brancato, R.; Ferrari, M.; Cremonesi, L. De novo deletion removes a conserved motif in the C-terminus of ABCA4 and results in cone-rod dystrophy. Clin. Chem. Lab. Med. 2006, 44, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Schubert, C.; Im, K.M.; Burke, T.; Brown, C.M.; Fishman, G.A.; Tsang, S.H.; Gouras, P.; Dean, M.; Allikmets, R. Analysis of the ABCA4 gene by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8479–8487. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Cremers, F.P.M.; Cornelis, S.S.; Runhart, E.H.; Astuti, G.D.N. Author Response: Penetrance of the ABCA4 p.Asn1868Ile Allele in Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5566–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, A.; Flothmann, K.; Hemmrich, N.; Ingvast, S.; Jorge, P.; Paloma, E.; Patel, R.; Rozet, J.M.; Tammur, J.; Testa, F.; et al. The ABCA4 2588G>C Stargardt mutation: Single origin and increasing frequency from South-West to North-East Europe. Eur. J. Hum. Genet. 2002, 10, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Aukrust, I.; Jansson, R.W.; Bredrup, C.; Rusaas, H.E.; Berland, S.; Jørgensen, A.; Haug, M.G.; Rødahl, E.; Houge, G.; Knappskog, P.M. The intronic ABCA4 c.5461-10T>C variant, frequently seen in patients with Stargardt disease, causes splice defects and reduced ABCA4 protein level. Acta Ophthalmol. 2017, 95, 240–246. [Google Scholar] [CrossRef]
- Midgley, N.; Roberts, L.; Rebello, G.; Ramesar, R. The impact of the c.5603A>T hypomorphic variant on founder mutation screening of ABCA4 for Stargardt disease in South Africa. Mol. Vis. 2020, 26, 613–622. [Google Scholar]
- Thompson, J.A.; Chiang, J.P.; De Roach, J.N.; McLaren, T.L.; Chen, F.K.; Hoffmann, L.; Campbell, I.; Lamey, T.M. Analysis of the ABCA4 c.[2588G>C;5603A>T] Allele in the Australian Population. Adv. Exp. Med. Biol 2019, 1185, 269–273. [Google Scholar] [CrossRef]
- Cremers, F.P.; Van de Pol, D.J.; Van Driel, M.; Den Hollander, A.I.; Van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Driel, M.A.; Maugeri, A.; Klevering, B.J.; Hoyng, C.B.; Cremers, F.P. ABCR unites what ophthalmologists divide(s). Ophthalmic Genet. 1998, 19, 117–122. [Google Scholar] [CrossRef]
- Shroyer, N.F.; Lewis, R.A.; Allikmets, R.; Singh, N.; Dean, M.; Leppert, M.; Lupski, J.R. The rod photoreceptor ATP-binding cassette transporter gene, ABCR, and retinal disease: From monogenic to multifactorial. Vis. Res. 1999, 39, 2537–2544. [Google Scholar] [CrossRef] [Green Version]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; Van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; Den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P. In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Oldani, M.; Marchi, S.; Giani, A.; Cecchin, S.; Rigoni, E.; Persi, A.; Podavini, D.; Guerrini, A.; Nervegna, A.; Staurenghi, G.; et al. Clinical and molecular genetic study of 12 Italian families with autosomal recessive Stargardt disease. Genet. Mol. Res. 2012, 11, 4342–4350. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Li, C.G.; Zhou, S.F. Prediction of deleterious functional effects of non-synonymous single nucleotide polymorphisms in human nuclear receptor genes using a bioinformatics approach. Drug Metab. Lett. 2009, 3, 242–286. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; Moore, A.T.; et al. A longitudinal study of Stargardt disease: Quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, R.W.; Ho, A.; Munoz, B.; Cideciyan, A.V.; Sahel, J.A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Studies: Design and Baseline Characteristics: ProgStar Report No. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Azarian, S.M.; Travis, G.H. The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt’s disease (ABCR). FEBS Lett. 1997, 409, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, F.; Burstedt, M.S.; Sandgren, O.; Norberg, A.; Golovleva, I. Novel mutations in CRB1 and ABCA4 genes cause Leber congenital amaurosis and Stargardt disease in a Swedish family. Eur. J. Hum. Genet. 2013, 21, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Garces, F.; Jiang, K.; Molday, L.L.; Stöhr, H.; Weber, B.H.; Lyons, C.J.; Maberley, D.; Molday, R.S. Correlating the Expression and Functional Activity of ABCA4 Disease Variants With the Phenotype of Patients With Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Curtis, S.B.; Molday, L.L.; Garces, F.A.; Molday, R.S. Functional analysis and classification of homozygous and hypomorphic ABCA4 variants associated with Stargardt macular degeneration. Hum. Mutat. 2020, 41, 1944–1956. [Google Scholar] [CrossRef]
- Garces, F.A.; Scortecci, J.F.; Molday, R.S. Functional Characterization of ABCA4 Missense Variants Linked to Stargardt Macular Degeneration. Int. J. Mol. Sci. 2020, 22, 185. [Google Scholar] [CrossRef]
- Radu, R.A.; Yuan, Q.; Hu, J.; Peng, J.H.; Lloyd, M.; Nusinowitz, S.; Bok, D.; Travis, G.H. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3821–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teussink, M.M.; Lee, M.D.; Smith, R.T.; Van Huet, R.A.C.; Klaver, C.C.; Klevering, B.J.; Theelen, T.; Hoyng, C.B. The Effect of Light Deprivation in Patients with Stargardt Disease. Am. J. Ophthalmol. 2015, 159, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wang, J.H.; Chen, J.; Li, F.; Edwards, T.L.; Hewitt, A.W.; Liu, G.S. Gene therapy for visual loss: Opportunities and concerns. Prog. Retin. Eye Res. 2019, 68, 31–53. [Google Scholar] [CrossRef]
- Campa, C.; Gallenga, C.E.; Bolletta, E.; Perri, P. The Role of Gene Therapy in the Treatment of Retinal Diseases: A Review. Curr. Gene Ther. 2017, 17, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P.M. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Moore, A.T.; Ali, R.R.; Bainbridge, J.W. Educational paper. Eur. J. Pediatrics 2012, 171, 757–765. [Google Scholar] [CrossRef]
- Oner, A. Recent Advancements in Gene Therapy for Hereditary Retinal Dystrophies. Turk. J. Ophthalmol. 2017, 47, 338–343. [Google Scholar] [CrossRef]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef]
- U.S Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (accessed on 1 June 2021).
- NICE. NICE Recommends Novel Gene Therapy Treatment for Rare Inherited Eye Disorder. Available online: https://www.nice.org.uk/news/article/nice-recommends-novel-gene-therapy-treatment-for-rare-inherited-eye-disorder (accessed on 1 June 2021).
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Issa, P.C.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4(-/-) Mice. Hum. Gene Ther. 2019, 30, 590–600. [Google Scholar] [CrossRef]
- Han, Z.; Conley, S.M.; Makkia, R.S.; Cooper, M.J.; Naash, M.I. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J. Clin. Investig. 2012, 122, 3221–3226. [Google Scholar] [CrossRef]
- Kong, J.; Kim, S.R.; Binley, K.; Pata, I.; Doi, K.; Mannik, J.; Zernant-Rajang, J.; Kan, O.; Iqball, S.; Naylor, S.; et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008, 15, 1311–1320. [Google Scholar] [CrossRef] [Green Version]
- Binley, K.; Widdowson, P.; Loader, J.; Kelleher, M.; Iqball, S.; Ferrige, G.; De Belin, J.; Carlucci, M.; Angell-Manning, D.; Hurst, F.; et al. Transduction of photoreceptors with equine infectious anemia virus lentiviral vectors: Safety and biodistribution of StarGen for Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4061–4071. [Google Scholar] [CrossRef]
- Dyka, F.M.; Molday, L.L.; Chiodo, V.A.; Molday, R.S.; Hauswirth, W.W. Dual ABCA4-AAV Vector Treatment Reduces Pathogenic Retinal A2E Accumulation in a Mouse Model of Autosomal Recessive Stargardt Disease. Hum. Gene Ther. 2019, 30, 1361–1370. [Google Scholar] [CrossRef]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; De Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef]
- Colella, P.; Trapani, I.; Cesi, G.; Sommella, A.; Manfredi, A.; Puppo, A.; Iodice, C.; Rossi, S.; Simonelli, F.; Giunti, M.; et al. Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 2014, 21, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Planul, A.; Dalkara, D. Vectors and Gene Delivery to the Retina. Annu. Rev. Vis. Sci. 2017, 3, 121–140. [Google Scholar] [CrossRef]
- Glover, D.J.; Lipps, H.J.; Jans, D.A. Towards safe, non-viral therapeutic gene expression in humans. Nat. Rev. Genet. 2005, 6, 299–310. [Google Scholar] [CrossRef]
- Sun, D.; Schur, R.M.; Sears, A.E.; Gao, S.-Q.; Vaidya, A.; Sun, W.; Maeda, A.; Kern, T.; Palczewski, K.; Lu, Z.-R. Non-viral Gene Therapy for Stargardt Disease with ECO/pRHO-ABCA4 Self-Assembled Nanoparticles. Mol. Ther. 2020, 28, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Conley, S.M.; Naash, M.I. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv. Exp. Med. Biol. 2014, 801, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.L.; Edwards, T.L.; O’Hare, F.; Hickey, D.G.; Wang, J.H.; Liu, Z.; Ayton, L.N. Gene therapy for inherited retinal diseases: Progress and possibilities. Clin. Exp. Optom. 2021, 104, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Semple-Rowland, S.L.; Berry, J. Use of lentiviral vectors to deliver and express bicistronic transgenes in developing chicken embryos. Methods 2014, 66, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyka, F.M.; Boye, S.L.; Chiodo, V.A.; Hauswirth, W.W.; Boye, S.E. Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 2014, 25, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Trapani, I.; Toriello, E.; De Simone, S.; Colella, P.; Iodice, C.; Polishchuk, E.V.; Sommella, A.; Colecchi, L.; Rossi, S.; Simonelli, F.; et al. Improved dual AAV vectors with reduced expression of truncated proteins are safe and effective in the retina of a mouse model of Stargardt disease. Hum. Mol. Genet. 2015, 24, 6811–6825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, P.C.; MacLaren, R.E. Non-viral retinal gene therapy: A review. Clin. Exp. Ophthalmol. 2012, 40, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Duijkers, L.; Van den Born, L.I.; Neidhardt, J.; Bax, N.M.; Pierrache, L.H.M.; Klevering, B.J.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Splicing Correction in Individuals with Leber Congenital Amaurosis due to Compound Heterozygosity for the c.2991+1655A>G Mutation in CEP290. Int. J. Mol. Sci. 2018, 19, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [Green Version]
- Bonifert, T.; Menendez, I.G.; Battke, F.; Theurer, Y.; Synofzik, M.; Schöls, L.; Wissinger, B. Antisense Oligonucleotide Mediated Splice Correction of a Deep Intronic Mutation in OPA1. Mol. Ther. Nucleic Acids 2016, 5, e390. [Google Scholar] [CrossRef] [Green Version]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef] [Green Version]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W.J. Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-Intronic ABCA4 Variant c.4539+2001G>A in Stargardt Disease. Genes 2019, 10, 452. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Arno, G.; Fakin, A.; Parfitt, D.A.; Dhooge, P.P.A.; Albert, S.; Bax, N.M.; Duijkers, L.; Niblock, M.; Hau, K.L.; et al. Detailed Phenotyping and Therapeutic Strategies for Intronic ABCA4 Variants in Stargardt Disease. Mol. Ther. Nucleic Acids 2020, 21, 412–427. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Song, W.K.; Park, K.-M.; Kim, H.-J.; Lee, J.H.; Choi, J.; Chong, S.Y.; Shim, S.H.; Del Priore, L.V.; Lanza, R. Treatment of macular degeneration using embryonic stem cell-derived retinal pigment epithelium: Preliminary results in Asian patients. Stem Cell Rep. 2015, 4, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Mehat, M.S.; Sundaram, V.; Ripamonti, C.; Robson, A.G.; Smith, A.J.; Borooah, S.; Robinson, M.; Rosenthal, A.N.; Innes, W.; Weleber, R.G.; et al. Transplantation of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells in Macular Degeneration. Ophthalmology 2018, 125, 1765–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.; Lee, M.J.; Choi, J.; Jung, S.Y.; Chong, S.Y.; Sung, J.H.; Shim, S.H.; Song, W.K. Long-term safety and tolerability of subretinal transplantation of embryonic stem cell-derived retinal pigment epithelium in Asian Stargardt disease patients. Br. J. Ophthalmol. 2020, 105, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Oner, A.; Gonen, Z.B.; Sevim, D.G.; Kahraman, N.S.; Unlu, M. Suprachoroidal Adipose Tissue-Derived Mesenchymal Stem Cell Implantation in Patients with Dry-Type Age-Related Macular Degeneration and Stargardt’s Macular Dystrophy: 6-Month Follow-Up Results of a Phase 2 Study. Cell. Reprogram. 2018, 20, 329–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, Y.; Ma, L.; Washington, I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J. Biol. Chem. 2011, 286, 7958–7965. [Google Scholar] [CrossRef] [Green Version]
- Issa, P.C.; Barnard, A.R.; Herrmann, P.; Washington, I.; MacLaren, R.E. Rescue of the Stargardt phenotype in Abca4 knockout mice through inhibition of vitamin A dimerization. Proc. Natl. Acad. Sci. USA 2015, 112, 8415–8420. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Kaufman, Y.; Zhang, J.; Washington, I. C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J. Biol. Chem. 2011, 286, 7966–7974. [Google Scholar] [CrossRef] [Green Version]
- Radu, R.A.; Han, Y.; Bui, T.V.; Nusinowitz, S.; Bok, D.; Lichter, J.; Widder, K.; Travis, G.H.; Mata, N.L. Reductions in Serum Vitamin A Arrest Accumulation of Toxic Retinal Fluorophores: A Potential Therapy for Treatment of Lipofuscin-Based Retinal Diseases. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4393–4401. [Google Scholar] [CrossRef] [PubMed]
- Dobri, N.; Qin, Q.; Kong, J.; Yamamoto, K.; Liu, Z.; Moiseyev, G.; Ma, J.-x.; Allikmets, R.; Sparrow, J.R.; Petrukhin, K. A1120, a Nonretinoid RBP4 Antagonist, Inhibits Formation of Cytotoxic Bisretinoids in the Animal Model of Enhanced Retinal Lipofuscinogenesis. Investig. Ophthalmol. Vis. Sci. 2013, 54, 85–95. [Google Scholar] [CrossRef]
- Bavik, C.; Henry, S.H.; Zhang, Y.; Mitts, K.; McGinn, T.; Budzynski, E.; Pashko, A.; Lieu, K.L.; Zhong, S.; Blumberg, B.; et al. Visual cycle modulation as an approach toward preservation of retinal integrity. PLoS ONE 2015, 10, e0124940. [Google Scholar] [CrossRef] [Green Version]
- Kubota, R.; Birch, D.G.; Gregory, J.K.; Koester, J.M. Randomised study evaluating the pharmacodynamics of emixustat hydrochloride in subjects with macular atrophy secondary to Stargardt disease. Br. J. Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Sieving, P.A.; Chaudhry, P.; Kondo, M.; Provenzano, M.; Wu, D.; Carlson, T.J.; Bush, R.A.; Thompson, D.A. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc. Natl. Acad. Sci. USA 2001, 98, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Mata, N.L.; Nusinowitz, S.; Liu, X.; Sieving, P.A.; Travis, G.H. Treatment with isotretinoin inhibits lipofuscin accumulation in a mouse model of recessive Stargardt’s macular degeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 4742–4747. [Google Scholar] [CrossRef] [Green Version]
- Piccardi, M.; Fadda, A.; Martelli, F.; Marangoni, D.; Magli, A.; Minnella, A.M.; Bertelli, M.; Di Marco, S.; Bisti, S.; Falsini, B. Antioxidant Saffron and Central Retinal Function in ABCA4-Related Stargardt Macular Dystrophy. Nutrients 2019, 11, 2461. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sabirzhanova, I.; Bergbower, E.A.S.; Yanda, M.; Guggino, W.G.; Cebotaru, L. The CFTR Corrector, VX-809 (Lumacaftor), Rescues ABCA4 Trafficking Mutants: A Potential Treatment for Stargardt Disease. Cell. Physiol. Biochem. 2019, 53, 400–412. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Golczak, M.; Chen, Y.; Okano, K.; Kohno, H.; Shiose, S.; Ishikawa, K.; Harte, W.; Palczewska, G.; Maeda, T.; et al. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol. 2011, 8, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Campagno, K.E.; Tso, H.Y.; Cenaj, A.; Laties, A.M.; Carlsson, L.G.; Mitchell, C.H. Oral Delivery of the P2Y12 Receptor Antagonist Ticagrelor Prevents Loss of Photoreceptors in an ABCA4-/- Mouse Model of Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3046–3053. [Google Scholar] [CrossRef]
- Lu, W.; Gómez, N.M.; Lim, J.C.; Guha, S.; O’Brien-Jenkins, A.; Coffey, E.E.; Campagno, K.E.; McCaughey, S.A.; Laties, A.M.; Carlsson, L.G.; et al. The P2Y(12) Receptor Antagonist Ticagrelor Reduces Lysosomal pH and Autofluorescence in Retinal Pigmented Epithelial Cells From the ABCA4-/- Mouse Model of Retinal Degeneration. Front. Pharmacol. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Schraermeyer, U. Lipofuscin can be eliminated from the retinal pigment epithelium of monkeys. Neurobiol. Aging 2012, 33, 2390–2397. [Google Scholar] [CrossRef] [PubMed]
- Julien-Schraermeyer, S.; Illing, B.; Tschulakow, A.; Taubitz, T.; Guezguez, J.; Burnet, M.; Schraermeyer, U. Penetration, distribution, and elimination of remofuscin/soraprazan in Stargardt mouse eyes following a single intravitreal injection using pharmacokinetics and transmission electron microscopic autoradiography: Implication for the local treatment of Stargardt’s disease and dry age-related macular degeneration. Pharmacol. Res. Perspect. 2020, 8, e00683. [Google Scholar] [CrossRef]
- Cao, S.; Wang, J.C.C.; Gao, J.; Wong, M.; To, E.; White, V.A.; Cui, J.Z.; Matsubara, J.A. CFH Y402H polymorphism and the complement activation product C5a: Effects on NF-κB activation and inflammasome gene regulation. Br. J. Ophthalmol. 2016, 100, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Csaky, K.G.; Bok, D.; Radu, R.A.; Sadda, S.R. Complement C5 Inhibition as a Potential Treatment for Autosomal Recessive Stargardt Disease (STGD1): Design of a Clinical Trial Assessing a Novel Treatment and Primary Outcome Measure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1569. [Google Scholar]
- MacDonald, I.M.; Sieving, P.A. Investigation of the effect of dietary docosahexaenoic acid (DHA) supplementation on macular function in subjects with autosomal recessive Stargardt macular dystrophy. Ophthalmic Genet. 2018, 39, 477–486. [Google Scholar] [CrossRef]
- Cornish, K.S.; Ho, J.; Downes, S.; Scott, N.W.; Bainbridge, J.; Lois, N. The Epidemiology of Stargardt Disease in the United Kingdom. Ophthalmol. Retin. 2017, 1, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Tsybovsky, Y.; Schwartz, S.B.; Windsor, E.A.M.; Roman, A.J.; Sumaroka, A.; Steinberg, J.D.; Jacobson, S.G.; et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet. 2009, 18, 931–941. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.; Chen, J. Molecular structures of the eukaryotic retinal importer ABCA4. Elife 2021, 10, e63524. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenocopy | Disease | Inheritance pattern | Associated phenotypes | References |
|---|---|---|---|---|
| ELOVL4 | STGD2 and STGD3 | Autosomal dominant | Macular pigmentary changes and flecks | [64,67] |
| PROM1 | STGD4 | Autosomal dominant | Cone-rod dystrophy Macular dystrophy Retinitis pigmentosa Bull’s eye maculopathy (BEM) Flecks | [32,65,68,69] |
| PRPH2 | Pattern dystrophy | Autosomal dominant | Pattern dystrophy Fleck like lesions that can be confused with STGD1. | [70,71] |
| CRX | Cone-rod dystrophy | Autosomal dominant | BEM | [72,73] |
| BEST1 | Bestrophinopathies | Autosomal recessive Autosomal dominant | Widespread vitelliform flecks in autosomal recessive Adult vitelliform lesion in autosomal dominant | [74,75] |
| CDH3 | Macular dystrophy | Autosomal recessive | Juvenile onset macular dystrophy with associated hyptrichosis of scalp hair | [76,77] |
| Hydroxychloroquine retinopathy | Bull’s eye maculopathy | Drug toxicity | Bull’s eye maculopathy | [58] |
| Population | ABCA4 Allele Detection Rate | Reference |
|---|---|---|
| Canada (isolated population Newfoundland) | 93% | [115] |
| Chinese | 61–84% | [103,116] |
| Polish | 79% | [117] |
| Danish | 77% | [99] |
| Portuguese | 76% | [100] |
| Spain | 76% | [98] |
| Germany | 74% | [118] |
| Mexico | 74% | [119] |
| USA | 50–75% | [120,121] |
| Hungarian | 65.7% | [122] |
| South Africa | 62% | [123] |
| Canadian | 59% | [116] |
| French Canadian | 33% | [116] |
| Description | Proportion of ProgStar cohort | Proportion in Adult Cohort with Age of Onset >17 Years [154] | Proportion in Paediatric Cohort with Age of Onset <17 years [154] | |
|---|---|---|---|---|
| A | Two or more severe or null variants | 5.7% | 1.6% | 20.6% |
| B | One severe/null variant and at least one missense or in frame deletion insertion | 44.4% | 40.6% | 44.1% |
| C | Two or more missense or in frame insertion/deletion variants | 49.8% | 54.7% | 35.2% |
| Class | Description | Pathogenicity |
|---|---|---|
| 1 | Truncating variant | Pathogenic |
| 2 | Non-truncating but enriched in ABCA4-LOVD data set compared to nFE ExAc control group | Likely pathogenic |
| 3 | Non-truncating variant that is more frequent in the ABCA4-LOVD dataset compared to ExAc control group but not significantly enriched | Uncertain significance |
| 4 | Variant had higher frequency in nFE ExAc control group than ABCA4-LOVD data set | Likely benign |
| 5 | Variant has a frequency > 0.005 in nFE ExAC population and not a known mild ABCA4 variant | Benign |
| Class | ABCA4 Expression | Basal ATPase Activity | Stimulation by N-Ret-PE |
|---|---|---|---|
| 1 | Significantly reduced | <50% | Not stimulated |
| 2 | Partial reduction | 50–80% | Modestly stimulated |
| 3 | Comparable to WT | Comparable to WT | Comparable to WT |
| Therapy | Mechanism of action | Result | Reference |
|---|---|---|---|
| Deuterated vitamin A | Vitamin A is deuterated at the carbon 20 position which strengthens the bond to the retinaldehyde-PE Schiff base which slows the production of A2E production and also provides more time for the Schiff base to be returned to the visual cycle | Abca4 KO Mice:
| [203,204,205] |
| RBP4 antagonists | Retinol binding protein 4 antagonists inhibit the binding of all-trans-retinol to RBP4 in the serum thus decreasing the transport of all-trans-retinol to the RPE and as a result decreasing bisretinoid production | Fenretidine in Abca4 KO:
| [206] [207] |
| Emixustat hydrochloride | Emixustat hydrochloride inhibits the RPE65 protein which reduces 11-cis-retinal thus all-trans-retinal and the subsequent A2E accumulation | Abca4 KO mice:
| [208] [209] |
| 4-methylpyrazole | Inhibits alcohol dehydrogenase | Humans
| |
| Isotretinoin | Inhibits 11-cis-retinol dehydrogenase in the RPE thus decreasing 11-cis-retinaldehyde production and rhodopsin regeneration | In Abca4 KO
| [210,211] |
| Saffron | Counteract oxidative damage through the carotenoid constituents crocins and crocetin | Shown to be safe in a double-blind placebo-controlled trial | [212] |
| VX-809 | Increase expression of ABCA4 protein in cells containing the p.(Ala1038Val) and p. (Gly1961Glu) variants | Increased expression of ABCA4 protein in HEK293T cells | [213] |
| Amine containing drugs | Sequesters all-trans-retinal by reacting with the aldehyde group and forming an inactive Schiff-base and those that compete with PE thus preventing A2E production | Abca4 KOPreserved retinal architecture in treated mice compared to untreated | [214] |
| Ticagrelor | Exposure to A2E increases the lysosomal pH which affects degeneration of the photoreceptors. Ticagrlor targets this by inhibiting the P2Y12 receptor to lower the lysosomal pH. | Abca4 KO
| [215,216] |
| Soraprazan | Reversible fast acting inhibitor H+, K+ ATPase that was noted to decrease lipofuscin deposits. | Decreased lipofuscin in RPE in treated monkeys and mice | [217,218] |
| Zimura® | Target C5 complement pathway to prevent formation of the membrane attack complex thus reducing cell death following activation of the complement pathway by A2E and bistretinoids | Results of Phase IIb study awaited | [219,220] |
| Omega 3 fatty acid supplementation | Omega 3 fatty acids are thought to be important for general retinal function | Trial results awaited | NCT03297515 |
| Docosahexaenoic acid (DHA) supplementation | DHA normally has a high concentration in the retina and is important towards retinal function | A trial showed no improvement in retinal function | [221] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Khuzaei, S.; Broadgate, S.; Foster, C.R.; Shah, M.; Yu, J.; Downes, S.M.; Halford, S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes 2021, 12, 1241. https://doi.org/10.3390/genes12081241
Al-Khuzaei S, Broadgate S, Foster CR, Shah M, Yu J, Downes SM, Halford S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes. 2021; 12(8):1241. https://doi.org/10.3390/genes12081241
Chicago/Turabian StyleAl-Khuzaei, Saoud, Suzanne Broadgate, Charlotte R. Foster, Mital Shah, Jing Yu, Susan M. Downes, and Stephanie Halford. 2021. "An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story" Genes 12, no. 8: 1241. https://doi.org/10.3390/genes12081241
APA StyleAl-Khuzaei, S., Broadgate, S., Foster, C. R., Shah, M., Yu, J., Downes, S. M., & Halford, S. (2021). An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes, 12(8), 1241. https://doi.org/10.3390/genes12081241