
Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations

 , , , ,
, , , ,
Abstract
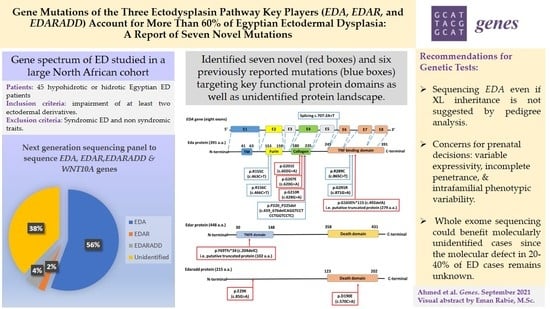
:
1. Introduction
2. Patients and Methods
2.1. Ethical Considerations
2.2. Clinical Evaluation
2.3. Molecular Investigation
2.3.1. Genomic DNA Extraction and Targeted NGS
2.3.2. Data Processing and Bioinformatic Analysis
2.3.3. Segregation Analyses
3. Results
3.1. Clinical Data of 28 Molecularly Identified ED Patients
3.2. Genetic Spectrum of ED in Our Cohort
3.2.1. EDA Mutations
3.2.2. Seven Novel Mutations and Their in Silico Analyses
3.2.3. Variable Expressivity of Heterozygous Carriers and De Novo Events
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. Part A 2019, 179, 442–447. [Google Scholar] [CrossRef]
- Pagnan, N.A.B.; Visinoni, Á.F. Update on ectodermal dysplasias clinical classification. Am. J. Med. Genet. Part A 2014, 164, 2415–2423. [Google Scholar] [CrossRef]
- Itin, P.H.; Fistarol, S.K. Ectodermal dysplasias. Am. J. Med. Genet. C Semin. Med. Genet. 2004, 131C, 45–51. [Google Scholar] [CrossRef]
- Fete, M.; Hermann, J.; Behrens, J.; Huttner, K.M. X-linked hypohidrotic ectodermal dysplasia (XLHED): Clinical and diagnostic insights from an international patient registry. Am. J. Med. Genet. Part A 2014, 164, 2437–2442. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, M.; Henningsen, E.; Hertz, J.M.; Vestergaard Grejsen, D.; Bygum, A. A retrospective study of clinical and mutational findings in 45 Danish families with ectodermal dysplasia. Acta Derm. Venereol. 2014, 94, 531–533. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, C.; Hadj-Rabia, S.; Jambou, M.; Mansour, S.; Guigue, P.; Masmoudi, S.; Bal, E.; Chassaing, N.; Vincent, M.-C.; Viot, G.; et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 2011, 32, 70–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavuz, I.; Baskan, Z.; Ulku, R.; Dulgergil, T.C.; Dari, O.; Ece, A.; Yavuz, Y.; Dari, K.O. Ectodermal Dysplasia: Retrospective Study of Fifteen Cases. Arch. Med. Res. 2006, 37, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Carlson, B.M. Genetic regulatory pathways of split-hand/foot malformation. Clin. Genet. 2019, 95, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koster, M.I. p63 in Skin Development and Ectodermal Dysplasias. J. Investig. Dermatol. 2010, 130, 2352–2358. [Google Scholar] [CrossRef] [Green Version]
- Vera-Carbonell, A.; Moya-Quiles, M.R.; Ballesta-Martínez, M.; López-González, V.; Bafallíu, J.A.; Guillén-Navarro, E.; López-Expósito, I. Rapp–Hodgkin syndrome and SHFM1 patients: Delineating the p63–Dlx5/Dlx6 pathway. Gene 2012, 497, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Nielsen, M.; Skovbo, S.; Svaneby, D.; Pedersen, L.; Fryzek, J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur. J. Med. Genet. 2013, 56, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Mortier, K.; Wackens, G. Ectodermal dysplasia, anhidrotic. Orphanet Encycl. 2004, 3, 1–6. [Google Scholar]
- Murdock, S.; Lee, J.Y.; Guckes, A.; Wright, J.T. A costs analysis of dental treatment for ectodermal dysplasia. J. Am. Dent. Assoc. 2005, 136, 1273–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfart, S.; Meiller, R.; Hammersen, J.; Park, J.; Menzel-Severing, J.; Melichar, V.O.; Huttner, K.; Johnson, R.; Porte, F.; Schneider, H. Natural history of X-linked hypohidrotic ectodermal dysplasia: A 5-year follow-up study. Orphanet J. Rare Dis. 2020, 15, 7. [Google Scholar] [CrossRef]
- Körber, I.; Klein, O.D.; Morhart, P.; Faschingbauer, F.; Grange, D.K.; Clarke, A.; Bodemer, C.; Maitz, S.; Huttner, K.; Kirby, N.; et al. Safety and immunogenicity of Fc-EDA, a recombinant ectodysplasin A1 replacement protein, in human subjects. Br. J. Clin. Pharmacol. 2020, 86, 2063–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, P.; Huttner, K.M.; Kirby, N.; Schneider, H. Intra-Amniotic Administration of EDI200 for the Treatment of Ectodermal Dysplasias. U.S. Patent 10,300,013, 28 May 2019. [Google Scholar]
- Mikkola, M.L. Molecular aspects of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. Part A 2009, 149A, 2031–2036. [Google Scholar] [CrossRef]
- Mikkola, M.L.; Thesleff, I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003, 14, 211–224. [Google Scholar] [CrossRef]
- Zhang, Y.; Tomann, P.; Andl, T.; Gallant, N.M.; Huelsken, J.; Jerchow, B.; Birchmeier, W.; Paus, R.; Piccolo, S.; Mikkola, M.L.; et al. Reciprocal Requirements for EDA/EDAR/NF-κB and Wnt/β-Catenin Signaling Pathways in Hair Follicle Induction. Dev. Cell 2009, 17, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Güven, Y.; Bal, E.; Altunoglu, U.; Yücel, E.; Hadj-Rabia, S.; Koruyucu, M.; Bahar Tuna, E.; Çıldır, Ş.; Aktören, O.; Bodemer, C.; et al. Turkish Ectodermal Dysplasia Cohort: From Phenotype to Genotype in 17 Families. Cytogenet. Genome Res. 2019, 157, 189–196. [Google Scholar] [CrossRef]
- Guazzarotti, L.; Tadini, G.; Mancini, G.E.; Sani, I.; Pisanelli, S.; Galderisi, F.; D’Auria, E.; Secondi, R.; Bottero, A.; Zuccotti, G. V WNT10A gene is the second molecular candidate in a cohort of young Italian subjects with ectodermal derivative impairment (EDI). Clin. Genet. 2018, 93, 693–698. [Google Scholar] [CrossRef]
- Martínez-Romero, M.C.; Ballesta-Martínez, M.J.; López-González, V.; Sánchez-Soler, M.J.; Serrano-Antón, A.T.; Barreda-Sánchez, M.; Rodriguez-Peña, L.; Martínez-Menchon, M.T.; Frías-Iniesta, J.; Sánchez-Pedreño, P.; et al. EDA, EDAR, EDARADD and WNT10A allelic variants in patients with ectodermal derivative impairment in the Spanish population. Orphanet J. Rare Dis. 2019, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Gaczkowska, A.; Abdalla, E.M.; Dowidar, K.M.L.; Elhady, G.M.; Jagodzinski, P.P.; Mostowska, A. De novo EDA mutations: Variable expression in two Egyptian families. Arch. Oral Biol. 2016, 68, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shawky, R.M.; Elsayed, N.S.; Ibrahim, D.S.; Seifeldin, N.S. Profile of genetic disorders prevalent in northeast region of Cairo, Egypt. Egypt. J. Med. Hum. Genet. 2012, 13, 45–62. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Monreal, A.W.; Zonana, J.; Ferguson, B. Identification of a New Splice Form of the EDA1 Gene Permits Detection of Nearly All X-Linked Hypohidrotic Ectodermal Dysplasia Mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Bayés, M.; Hartung, A.J.; Ezer, S.; Pispa, J.; Thesleff, I.; Srivastava, A.K.; Kere, J. The Anhidrotic Ectodermal Dysplasia Gene (EDA) Undergoes Alternative Splicing and Encodes Ectodysplasin-A with Deletion Mutations in Collagenous Repeats. Hum. Mol. Genet. 1998, 7, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Hammersen, J.; Preisler-Adams, S.; Huttner, K.; Rascher, W.; Bohring, A. Sweating ability and genotype in individuals with X-linked hypohidrotic ectodermal dysplasia. J. Med. Genet. 2011, 48, 426. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Han, D.; Qu, H.; Gong, Y.; Wu, H.; Zhang, X.; Zhong, N.; Feng, H. EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 2009, 88, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements. J. Hum. Genet. 2016, 61, 891–897. [Google Scholar] [CrossRef]
- Callea, M.; Vinciguerra, A.; Willoughby, C.E.; Deroma, L.; Clarich, G. Infantile bilateral glaucoma in a child with ectodermal dysplasia. Ophthalmic Genet. 2013, 34, 58–60. [Google Scholar] [CrossRef]
- Burger, K.; Schneider, A.-T.; Wohlfart, S.; Kiesewetter, F.; Huttner, K.; Johnson, R.; Schneider, H. Genotype–phenotype correlation in boys with X-linked hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. Part A 2014, 164, 2424–2432. [Google Scholar] [CrossRef]
- He, H.; Han, D.; Feng, H.; Qu, H.; Song, S.; Bai, B.; Zhang, Z. Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS ONE 2013, 8, e80393. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.I.; Wang, W.J.; Zhu, R.F.; Zhu, X.Y.; Yang, Y.; Wu, X. Molecular genetics study of ED1 gene for two X-linked hypohidrotic ectodermal dysplasia families. Chin. J. Med. Genet. 2013, 30, 399–402. [Google Scholar]
- Hymowitz, S.G.; Compaan, D.M.; Yan, M.; Wallweber, H.J.A.; Dixit, V.M.; Starovasnik, M.A.; de Vos, A.M. The Crystal Structures of EDA-A1 and EDA-A2: Splice Variants with Distinct Receptor Specificity. Structure 2003, 11, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Shen, W.; Wang, Y.; Liu, Y.; Liu, H.; Zhao, H.; Zhang, G.; Snead, M.L.; Han, D.; Feng, H. Functional Study of Ectodysplasin-A Mutations Causing Non-Syndromic Tooth Agenesis. PLoS ONE 2016, 11, e0154884. [Google Scholar] [CrossRef] [Green Version]
- Casal, M.L.; Lewis, J.R.; Mauldin, E.A.; Tardivel, A.; Ingold, K.; Favre, M.; Paradies, F.; Demotz, S.; Gaide, O.; Schneider, P. Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am. J. Hum. Genet. 2007, 81, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Heiland, G.; Jabir, S.; Wende, W.; Blecher, S.; Bock, N.; Ruf, S. Novel missense mutation in the EDA gene in a family affected by oligodontia. J. Orofac. Orthop. 2016, 77, 31–38. [Google Scholar] [CrossRef]
- Bock, N.C.; Lenz, S.; Ruiz-Heiland, G.; Ruf, S. Nonsyndromic oligodontia. J. Orofac. Orthop. 2017, 78, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Ko, J.; Shin, T.J.; Hyun, H.K.; Lee, S.H.; Kim, J.W. Oligodontia and Curly Hair Occur with Ectodysplasin-A Mutations. J. Dent. Res. 2014, 93, 371–375. [Google Scholar] [CrossRef]
- Park, J.S.; Ko, J.M.; Chae, J.-H. Novel and Private EDA Mutations and Clinical Phenotypes of Korean Patients with X-Linked Hypohidrotic Ectodermal Dysplasia. Cytogenet. Genome Res. 2019, 158, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van der Hout, A.H.; Oudesluijs, G.G.; Venema, A.; Verheij, J.B.G.M.; Mol, B.G.J.; Rump, P.; Brunner, H.G.; Vos, Y.J.; van Essen, A.J. Mutation screening of the Ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 2008, 16, 673–679. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene-Exon Number | Forward (F) and Reverse (R) Primers | Amplicon Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| EDA-2 | F 5′-TACAGTGGAGGGGAAGATGG-3′ | 388 | 61 |
| R 5′-ACCATGCCCTACCAAGAAGG-3′ | |||
| EDA-4 | F 5′-CTGGGCAACAGAGCAGG-3′ | 306 | 60 |
| R 5′-CCCACTCCTGCTCTCCTAAAG-3′ | |||
| EDA-7 | F 5′-AAAGTTTGGCCTTCTAGGCTAC-3′ | 418 | 60 |
| R 5′-CTTTCAACTCCCTCCCAGTG-3′ | |||
| EDAR-4 | F 5′-CATCTGGAGCCTGAGAGTGG-3′ | 495 | 61 |
| R 5′-GCAGTATCCATGACCCCTGTT-3′ | |||
| EDARADD-2 | F 5′-CTACCTCACCCAGCCAATCC-3′ | 466 | 62 |
| R 5′-CACCTCCAACATGAGCAAAAGA-3′ | |||
| EDARADD-6 | F 5′-CGAGCATTCTGAAATAGTCTTCC-3′ | 619 | 60 |
| R 5′-CTGTTCCACGTCCTTGTCCT-3′ |
| Family | Patient | Consanguinity | Sex | Age | Sweating | Hair | Skin | Nails | Degree of Missing Teeth | Peg Shaped Teeth |
|---|---|---|---|---|---|---|---|---|---|---|
| F1 | ED1 | − | M | 7 y 1 m | H | Sparse | Dry | N | O | + |
| F2 | ED2 | − | M | 12 y | H | Sparse | Dry | N | O | + |
| F2 | ED3 | − | M | 7 y | N | Sparse | Dry | N | O | + |
| F3 | ED4 | − | M | 3 y 5 m | H | Sparse | Dry | N | O | + |
| F4 | ED5 | − | F | 11 y | H | Sparse | Dry | N | O | + |
| F4 | ED6 | − | M | 48 y | H | Sparse | Dry | Dysplastic | O | + |
| F4 | ED7 | − | F | 4 y | N | Sparse | Dry | N | O | + |
| F4 | ED8 | − | M | 45 y | H | Sparse | Dry | Dysplastic | O | − |
| F4 | ED9 | − | F | 6 y 7 m | N | Sparse | Dry | N | O | + |
| F4 | ED10 | − | M | 59 y | H | Sparse | Dry | Dysplastic | O | − |
| F5 | ED11 | + | F | 9 y | H | Sparse | Dry | N | O | − |
| F6 | ED12 | − | M | 13 m | H | Sparse | Dry, thin | N | N/A | + |
| F7 | ED13 | − | M | 1 y 9 m | H | Fair, Sparse | Dry | N | N/A | − |
| F8 | ED14 | − | F | 3.5 y | H | Sparse | Dry | Dysplastic | N/A | + |
| F8 | ED15 | − | F | 1.5 y | N | Sparse | Dry | N | N/A | + |
| F9 | ED16 | + | M | 7 y | H | Sparse | Dry | N | O | + |
| F10 | ED17 | + | M | 4 y 7 m | H | Sparse | Dry | N | N/A | + |
| F11 | ED18 | − | M | 2 y 3 m | H | Fair, Sparse | - | N | N/A | − |
| F12 | ED19 | + | M | 20 y | H | Sparse | Dry | Dysplastic | O | + |
| F12 | ED20 | + | M | 24 y | H | Sparse | Dry | Dysplastic | O | + |
| F13 | ED21 | + | M | 11 y | H | Sparse | Dry | N | O | + |
| F14 | ED22 | − | M | 9 y | H | Sparse | Dry | N | O | + |
| F15 | ED23 | − | M | 2 y | N | Sparse | - | N | N/A | + |
| F16 | ED24 | − | F | 10 y | H | Sparse | Slightly dry | N | O | + |
| F17 | ED25 | + | M | 6 y | H | Sparse | Dry, thin | N | O | + |
| F18 | ED26 | + | M | 5 y | H | Silky, Sparse | - | N | N/A | + |
| F19 | ED27 | + | M | 3 y 10 m | H | Sparse | Dry | N | N/A | − |
| F20 | ED28 | + | F | 3 y | H | Sparse | - | N | N/A | − |
| Family | Patient | Gene | Exon (E) or Intron (IVS) | Mutation | Type of Mutation | Genotype | Variant Effect ** | Mode of Inheritance *** | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Nucleotide Change * | Protein Change | |||||||||
| F1–F4 | ED1–ED10 | EDA | E2 | c.463C > T | p.R155C | Missense | ED1-4,6,8,10: Hemizygous ED5,7,9: Heterozygous | Pathogenic | XL | [38] |
| F5–F7 | ED11–ED13 | EDA | E2 | c.466C > T | p.R156C | Missense | ED11: Heterozygous ED12,13: Hemizygous | Pathogenic | XL | [38] |
| F8 | ED14 and ED15 | EDA | E2 | c.492delA | p.G165Efs*115 | Small deletion | Heterozygous | Pathogenic | XL | Current study |
| F9–10 | ED16 and ED17 | EDA | E4 | c.602G > A | p.G201E | Missense | Hemizygous | Pathogenic | XL | Current study |
| F11 | ED18 | EDA | E4 | c.620G > A | p.G207E | Missense | Hemizygous | Pathogenic | XL | Current study |
| F12 | ED19 and ED20 | EDA | E4 | c.628G > A | p.G210R | Missense | Hemizygous | Pathogenic | XL | Current study |
| F13 | ED21 | EDA | E4 | c.659_676delCAGGTCCTCCTGGTCCTC | p.P220_P225del | Small deletion | Hemizygous | Pathogenic | De novo | [39] |
| F14 | ED22 | EDA | IVS5 | c.707-2A > T | p.? | Splicing | Hemizygous | Pathogenic | XL | [40] |
| F15 | ED23 | EDA | E7 | c.865C > T | p.R289C | Missense | Hemizygous | Pathogenic | XL | [41] |
| F16–17 | ED24 and ED25 | EDA | E7 | c.871G > A | p.G291R | Missense | ED24: Heterozygous ED25: Hemizygous | Pathogenic | ED24: De novo ED25: XL | [39] |
| F18 | ED26 | EDARADD | E2 | c.85G > A | p.E29K | Missense | Homozygous | Pathogenic | AR | Current study |
| F19 | ED27 | EDARADD | E6 | c.570C > A | p.D190E | Missense | Homozygous | Pathogenic | AR | Current study |
| F20 | ED28 | EDAR | E4 | c.204delC | p.Y69Tfs*34 | Small deletion | Homozygous | Pathogenic | AR | Current study |
| Gene | Novel Mutation | SIFT | PhD-SNP | Mutation Assessor | Polphen-2 | Mutation Taster | PROVEAN | SNPs&GO | CADD Score |
|---|---|---|---|---|---|---|---|---|---|
| EDA | c.602G > A (p.G201E) | Not tolerated/Damaging (0.00) | Disease (RI = 2) | Medium (3.2) | Probably damaging (1) | Disease causing (Score = 98) | Deleterious (−4.2) | Disease (RI = 8) | 26.3 |
| EDA | c.620G > A (p.G207E) | Not tolerated/Damaging (0.00) | Disease (RI = 2) | Medium (3.335) | Probably damaging (1) | Disease causing (Score = 98) | Deleterious (−4.185) | Disease (RI = 1) | 26.4 |
| EDA | c.628G > A (p.G210R) | Not tolerated/Damaging (0.00) | Disease (RI = 4) | Medium (3.495) | Probably damaging (0.981) | Disease causing (Score = 125) | Deleterious (−2.55) | Disease (RI = 9) | 26.0 |
| EDARADD | c.85G > A (p.E29K) | Not tolerated/Damaging (0.00) | Disease (RI = 2) | Medium (2.085) | Probably damaging (0.978) | Disease causing (Score = 56) | Neutral (−2.06) | Disease (RI = 8) | 28.9 |
| EDARADD | c.570C > A (p.D190E) | Not tolerated/Damaging (0.00) | Disease (RI = 4) | Low (1.04) | Probably damaging (0.999) | Disease causing (Score = 45) | Deleterious (−2.97) | Disease (RI = 9) | 23.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, H.A.; El-Kamah, G.Y.; Rabie, E.; Mostafa, M.I.; Abouzaid, M.R.; Hassib, N.F.; Mehrez, M.I.; Abdel-Kader, M.A.; Mohsen, Y.H.; Zada, S.K.; et al. Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations. Genes 2021, 12, 1389. https://doi.org/10.3390/genes12091389
Ahmed HA, El-Kamah GY, Rabie E, Mostafa MI, Abouzaid MR, Hassib NF, Mehrez MI, Abdel-Kader MA, Mohsen YH, Zada SK, et al. Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations. Genes. 2021; 12(9):1389. https://doi.org/10.3390/genes12091389
Chicago/Turabian StyleAhmed, Hoda A., Ghada Y. El-Kamah, Eman Rabie, Mostafa I. Mostafa, Maha R. Abouzaid, Nehal F. Hassib, Mennat I. Mehrez, Mohamed A. Abdel-Kader, Yasmine H. Mohsen, Suher K. Zada, and et al. 2021. "Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations" Genes 12, no. 9: 1389. https://doi.org/10.3390/genes12091389
APA StyleAhmed, H. A., El-Kamah, G. Y., Rabie, E., Mostafa, M. I., Abouzaid, M. R., Hassib, N. F., Mehrez, M. I., Abdel-Kader, M. A., Mohsen, Y. H., Zada, S. K., Amr, K. S., & Sayed, I. S. M. (2021). Gene Mutations of the Three Ectodysplasin Pathway Key Players (EDA, EDAR, and EDARADD) Account for More than 60% of Egyptian Ectodermal Dysplasia: A Report of Seven Novel Mutations. Genes, 12(9), 1389. https://doi.org/10.3390/genes12091389