
Identification of Two Novel EPOR Gene Variants in Primary Familial Polycythemia: Case Report and Literature Review

 ,
,  , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mcmullin, M.F. Genetic background of congenital erythrocytosis. Genes 2021, 12, 1151. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Ferrari, A.; Vannucchi, A.M.; Tefferi, A. The new WHO classification for essential thrombocythemia calls for revision of available evidences. Blood Cancer J. 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Prchal, J.T. Genetic heterogeneity of primary familial and congenital polycythemia. Am. J. Hematol. 2001, 68, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.; McMullin, M.F.; Gardie, B.; Percy, M.; Cario, H.; Kučerová, J.; Girodon, F.; Rossi, C.; Aström, M.; Diaz-Aguado, A.; et al. Congenital Erythrocytosis and Hereditary Thrombocytosis; COST (European Cooperation in Science and Technology): Brussels, Belgium, 2015; ISBN 978-989-20-5646-3. [Google Scholar]
- Bento, C.; Percy, M.J.; Gardie, B.; Maia, T.M.; van Wijk, R.; Perrotta, S.; Della Ragione, F.; Almeida, H.; Rossi, C.; Girodon, F.; et al. Genetic basis of congenital erythrocytosis: Mutation update and online databases. Hum. Mutat. 2014, 35, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.-S.; Shen, Y.-M.; Bulut, G.B. Advances in understanding the pathogenesis of primary familial and congenital polycythaemia. Br. J. Haematol. 2010, 101, 1306–1318. [Google Scholar] [CrossRef]
- Remy, I.; Wilson, I.A.; Michnick, S.W. Erythropoietin receptor activation by a ligand-induced conformation change. Science 1999, 283, 990–993. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Florensa, L.; Besses, C.; Woessner, S.; Solé, F.; Acín, P.; Pedro, C.; Sans-Sabrafen, J. Endogenous megakaryocyte and erythroid colony formation from blood in essential thrombocythaemia. Leukemia 1995, 9, 271–273. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Peroni, E.; Bertozzi, I.; Gherlinzoni, F.; Stefani, P.M.; Lombardi, A.; Biagetti, G.; Fabris, F.; Randi, M.L. Two novel missense mutations in EPOR gene causes erythrocytosis in two unrelated patients. Br. J. Haematol. 2018, 180, 908–911. [Google Scholar] [CrossRef]
- Bento, C.; Almeida, H.; Fernandez-Lago, C.; Ribeiro, M.L. Primary familial congenital erythrocytosis: Two novel EPOR mutations found in Spain. Int. J. Lab. Hematol. 2013, 35, e27–e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loganathan, S.E.; Kattaru, S.; Chandrasekhar, C.; Vengamma, B.; Sarma, P.V.G.K. Novel mutations in EPO-R and oxygen-dependent degradation (ODD) domain of EPAS1 genes-a causative reason for Congenital Erythrocytosis. Eur. J. Med. Genet. 2022, 65, 1–20. [Google Scholar] [CrossRef]
- Al-Sheikh, M.; Mazurier, E.; Gardie, B.; Casadevall, N.; Galactéros, F.; Goossens, M.; Wajcman, H.; Préhu, C.; Ugo, V. A study of 36 unrelated cases with pure erythrocytosis revealed three new mutations in the erythropoietin receptor gene. Haematologica 2008, 93, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Arcasoy, M.; Karayal, A.; Segal, H.; Sinning, J.; Forget, B. A novel mutation in the erythropoietin receptor gene is associated with familial erythrocytosis. Blood 2002, 99, 3066–3069. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, N.; Kaneda, M.; Hatakeyama, N.; Manabe, H.; Okajima, K.; Sakurai, Y.; Yamamoto, M.; Sarashina, T.; Ikuta, K.; Azuma, H. A case of primary familial congenital polycythemia with a novel EPOR mutation: Possible spontaneous remission/alleviation by menstrual bleeding. Int. J. Hematol. 2018, 108, 339–343. [Google Scholar] [CrossRef]
- O’Rourke, K.; Fairbairn, D.J.; Jackson, K.A.; Morris, K.L.; Tey, S.K.; Kennedy, G.A. A novel mutation of the erythropoietin receptor gene associated with primary familial and congenital polycythaemia. Int. J. Hematol. 2011, 93, 542–544. [Google Scholar] [CrossRef]
- Gross, M.; Ben-Califa, N.; McMullin, M.F.; Percy, M.J.; Bento, C.; Cario, H.; Minkov, M.; Neumann, D. Polycythaemia-inducing mutations in the erythropoietin receptor (EPOR): Mechanism and function as elucidated by epidermal growth factor receptor-EPOR chimeras. Br. J. Haematol. 2014, 165, 519–528. [Google Scholar] [CrossRef]
- Perrotta, S.; Cucciolla, V.; Ferraro, M.; Ronzoni, L.; Tramontano, A.; Rossi, F.; Scudieri, A.C.; Borriello, A.; Roberti, D.; Nobili, B.; et al. EPO receptor gain-of-function causes hereditary polycythemia, alters CD34+ cell differentiation and increases circulating endothelial precursors. PLoS ONE 2010, 5, e12015. [Google Scholar] [CrossRef]
- Petersen, K.B.; Hokland, P.; Petersen, G.B.; Nyvold, C.G. Erythropoietin receptor defect: A cause of primary polycythaemia. Br. J. Haematol. 2004, 125, 537–538. [Google Scholar] [CrossRef]
- Kralovics, R.; Sokol, L.; Prchal, J.T. Absence of polycythemia in a child with a unique erythropoietin receptor mutation in a family with autosomal dominant primary polycythemia. J. Clin. Investig. 1998, 102, 124–129. [Google Scholar] [CrossRef]
- Rives, S.; Pahl, H.L.; Florensa, L.; Bellosillo, B.; Neusuess, A.; Estella, J.; Debatin, K.M.; Kohne, E.; Schwarz, K.; Cario, H. Molecular genetic analyses in familial and sporadic congenitalprimary erythrocytosis. Haematologica 2007, 92, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Indrak, K.; Stopka, T.; Berman, B.W.; Prchal, J.F.; Prchal, J.T. Two new EPO receptor mutations: Truncated EPO receptors are most frequently associated with primary familial and congenital polycythemias. Blood 1997, 90, 2057–2061. [Google Scholar] [CrossRef] [PubMed]
- Watowich, S.; Xie, X.; Klingmüller, U.; Kere, J.; Lindlof, M.; Berglund, S.; De La Chapelle, A. Erythropoietin receptor mutations associated with familial erythrocytosis cause hypersensitivity to erythropoietin in the heterozygous state. Blood 1999, 94, 2530–2532. [Google Scholar] [CrossRef] [PubMed]
- Sokol, L.; Luhovy, M.; Guan, Y.; Prchal, J.F.; Semenza, G.L.; Prchal, J.T. Primary familial polycythemia: A frameshift mutation in the erythropoietin receptor gene and increased sensitivity of erythroid progenitors to erythropoietin. Blood 1995, 86, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Filser, M.; Aral, B.; Airaud, F.; Chauveau, A.; Bruce, A.; Polfrit, Y.; Thiebaut, A.; Gauthier, M.; le Marechal, C.; Lippert, E.; et al. Low incidence of EPOR mutations in idiopathic erythrocytosis. Haematologica 2021, 106, 299–301. [Google Scholar] [CrossRef]
- Furukawa, T.; Narita, M.; Sakaue, M.; Otsuka, T.; Kuroha, T.; Masuko, M.; Azegami, T.; Kishi, K.; Takahashi, M.; Utsumi, J.; et al. Primary familial polycythaemia associated with a novel point mutation in the erythropoietin receptor. Br. J. Haematol. 1997, 99, 222–227. [Google Scholar] [CrossRef]
- Pasquier, F.; Marty, C.; Balligand, T.; Verdier, F.; Grosjean, S.; Gryshkova, V.; Raslova, H.; Constantinescu, S.N.; Casadevall, N.; Vainchenker, W.; et al. New pathogenic mechanisms induced by germline erythropoietin receptor mutations in primary erythrocytosis. Haematologica 2017, 103, 575–586. [Google Scholar] [CrossRef]
- Chauveau, A.; Luque, D.; Lecucq, L.; Le Gac, G.; Le Maréchal, C.; Gueguen, P.; Berthou, C.; Ugo, V. A new point mutation in EPOR inducing a short deletion incongenital erythrocytosis. Br. J. Haematol. 2015, 172, 461–486. [Google Scholar] [CrossRef]
- Bento, C.; Almeida, H.; Maia, T.M.; Relvas, L.; Oliveira, A.C.; Rossi, C.; Girodon, F.; Fernandez-Lago, C.; Aguado-Diaz, A.; Fraga, C.; et al. Molecular study of congenital erythrocytosis in 70 unrelated patients revealed a potential causal mutation in less than half of the cases (Where is/are the missing gene(s)?). Eur. J. Haematol. 2013, 91, 361–368. [Google Scholar] [CrossRef]
- De la Chapelle, A.; Traskelin, A.L.; Juvonen, E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc. Natl. Acad. Sci. USA 1993, 90, 4495–4499. [Google Scholar] [CrossRef]
- Percy, M.J.; McMullin, M.F.; Roques, A.W.; Westwood, N.B.; Acharya, J.; Hughes, A.E.; Lappin, T.R.J.; Pearson, T.C. Erythrocytosis due to a mutation in the erythropoietin receptor gene. Br. J. Haematol. 1998, 100, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Le Couedic, J.; Mitjavila, M.; Villeval, J.-L.; Feger, F.; Gobert, S.; Mayeux, P.; Casadevall, N.; Vainchenker, W. Missense Mutation of the Erythropoietin Receptor Is a Rare Event in Human Erythroid Malignancies. Blood 1996, 87, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Aljabry, M. Primary familial and congenital polycythemia; The forgotten entity. J. Appl. Hematol. 2018, 9, 39. [Google Scholar] [CrossRef]
- Bento, C.; McMullin, M.F.; Percy, M.; Cario, H.; Adam, M.; Everman, D.; Mirzaa, G.; Pagon, R.; Wallace, S.; Bean, L.; et al. Primary Familial and Congenital Polycythemia. In GeneReviews [Internet]; University of Washington: Seattle WA, USA, 2016. [Google Scholar] [PubMed]
- Camps, C.; Petousi, N.; Bento, C.; Cario, H.; Copley, R.R.; McMullin, M.F.; Van Wijk, R.; Ratcliffe, P.; Robbins, P.A.; Taylor, J.C.; et al. Gene panel sequencing improves the diagnostic work-up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica 2016, 101, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Juvonen, E.; Ikkala, E.; Fyhrquist, F.; Ruutu, T. Autosomal Dominant Erythrocytosis Caused by Increased Sensitivity to Erythropoietin. Blood 1991, 78, 3066–3069. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, C.; Mayeux, P. Erythropoietin (Epo) receptor and Epo mimetics. Adv. Nephrol. Necker. Hosp. 1999, 29, 177–189. [Google Scholar]
- Bittorf, T.; Seiler, J.; Zhang, Z.; Jaster, R.; Brock, J. SHP1 protein tyrosine phosphatase negatively modulates erythroid differentiation and suppression of apoptosis in J2E erythroleukemic cells. Biol. Chem. 1999, 380, 1201–1209. [Google Scholar] [CrossRef]


{kind=link}
| Exon | Nucleotide Change | Protein Effect | Variant Type | References | Class |
|---|---|---|---|---|---|
| 8 | c.1013G>A | p.(Cys338Tyr) | Missense | [11] | VUS |
| 8 | c.1022C>T | p.(Thr341Met) | Missense | [11] | VUS |
| 8 | c.1138C>G | p.(Pro380Ala) | Missense | [12] | B |
| 8 | c.1183G>C | p.(Val395Leu) | Missense | [13] | LB |
| 8 | c.1142_1143del | p.(Pro381Glnfs∗2) | Frameshift | [14] | PAT |
| 8 | c.1195G>T | p.(Glu399∗) | Nonsense | [15] | PAT |
| 8 | c.1220C>A | p.(Ser407∗) | Nonsense | [16] | LPAT |
| 8 | c.1228A>C | p.(Gln343Pro) | Missense | [13] | VUS |
| 8 | c.1234del | p.(Ser412Argfs∗41) | Frameshift | [17] | PAT |
| 8 | c.1235C>A | p.(Ser412∗) | Nonsense | [12,18] | PAT |
| 8 | c.1242_1276del | p.(Ser415Hisfs∗18) | Frameshift | [12,18] | PAT |
| 8 | c.1249G>T | p.(Glu417∗) | Nonsense | [19] | PAT |
| 8 | c.1252 1255del | p.(Gly418Profs∗34) | Frameshift | [20] | PAT |
| 8 | c.1271_1272del | p.(Phe424∗) | Nonsense | [14] | PAT |
| 8 | c.1273G>T | p.(Glu425∗) | Nonsense | [3] | LPAT |
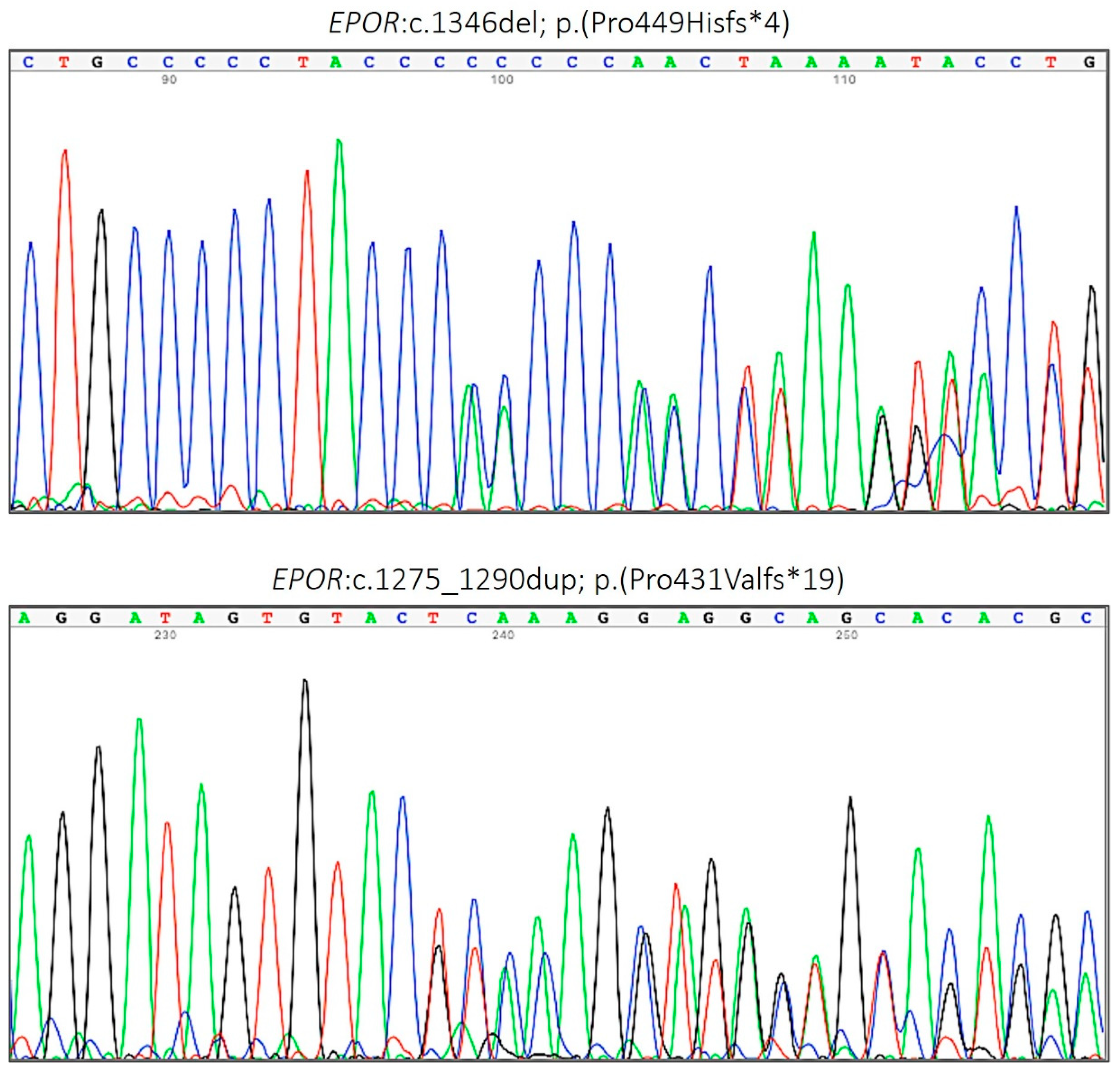
| 8 | c.1275_1290dup | p.(Pro431Valfs*19) | Frameshift | Present study | PAT |
| 8 | c.1278C>G | p.(Tyr426∗) | Nonsense | [21,22] | PAT |
| 8 | c.1281dup | p.(Ile428Tyrfs∗17) | Frameshift | [23] | PAT |
| 8 | c.1283_1289dup | p.(Ser432Glyfs*15) | Frameshift | [24] | PAT |
| 8 | c.1285del | p.(Leu429Trpfs∗24) | Frameshift | [14] | PAT |
| 8 | c.1288dup | p.(Asp430Glyfs∗15) | Frameshift | [25] | PAT |
| 8 | c.1293del | p.(Ser432Alafs∗21) | Frameshift | [26] | PAT |
| 8 | c.1299_1305del | p.(Gln434Cysfs∗17) | Frameshift | [15,23] | PAT |
| 8 | c.1300C>T | p.(Gln434∗) | Nonsense | [27] | PAT |
| 8 | c.1300dup | p.(Gln434Profs*11) | Frameshift | [28] | PAT |
| 8 | c.1310G>A | p.(Arg437His) | Missense | [12,18,29] | LB |
| 8 | c.1311_1312del | p.(Pro438Metfs∗6) | Frameshift | [30] | PAT |
| 8 | c.1316G>A | p.(Trp439∗) | Nonsense | [31,32] | PAT |
| 8 | c.1317G>A | p.(Trp439∗) | Nonsense | [22] | PAT |
| 8 | c.1346del | p.(Pro449Hisfs∗4) | Frameshift | Present study | PAT |
| 8 | c.1362C>G | p.(Tyr454∗) | Nonsense | [29] | LPAT |
| 8 | c.1460A>G | p.(Asn487Ser) | Missense | [14,33] | LB |
| 8 | c.1462C>T | p.(Pro488Ser) | Missense | [23,25] | LPAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo Riso, L.; Vargas-Parra, G.; Navarro, G.; Arenillas, L.; Fernández-Ibarrondo, L.; Robredo, B.; Ballester, C.; López, B.; Perez-Montaña, A.; Sampol, A.; et al. Identification of Two Novel EPOR Gene Variants in Primary Familial Polycythemia: Case Report and Literature Review. Genes 2022, 13, 1686. https://doi.org/10.3390/genes13101686
Lo Riso L, Vargas-Parra G, Navarro G, Arenillas L, Fernández-Ibarrondo L, Robredo B, Ballester C, López B, Perez-Montaña A, Sampol A, et al. Identification of Two Novel EPOR Gene Variants in Primary Familial Polycythemia: Case Report and Literature Review. Genes. 2022; 13(10):1686. https://doi.org/10.3390/genes13101686
Chicago/Turabian StyleLo Riso, Laura, Gardenia Vargas-Parra, Gemma Navarro, Leonor Arenillas, Lierni Fernández-Ibarrondo, Beatriz Robredo, Carmen Ballester, Bernardo López, Albert Perez-Montaña, Antonia Sampol, and et al. 2022. "Identification of Two Novel EPOR Gene Variants in Primary Familial Polycythemia: Case Report and Literature Review" Genes 13, no. 10: 1686. https://doi.org/10.3390/genes13101686
APA StyleLo Riso, L., Vargas-Parra, G., Navarro, G., Arenillas, L., Fernández-Ibarrondo, L., Robredo, B., Ballester, C., López, B., Perez-Montaña, A., Sampol, A., Florensa, L., Besses, C., Duran, M. A., & Bellosillo, B. (2022). Identification of Two Novel EPOR Gene Variants in Primary Familial Polycythemia: Case Report and Literature Review. Genes, 13(10), 1686. https://doi.org/10.3390/genes13101686