

Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
2.2. Overview of the GHTNet Architecture
2.2.1. Encoding Module
2.2.2. Pseudo Siamese C-Transformer Module
2.2.3. Pseudo Siamese CNN Module
2.3. Model Training
2.3.1. Word2vec Model Training
2.3.2. GHTNet Training
2.4. Comparison with Five TF-DNA Binding Prediction Methods
3. Results
3.1. Model Validation
3.1.1. GHTNet Achieves Superior Performance by Integrating Different Chromatin Features
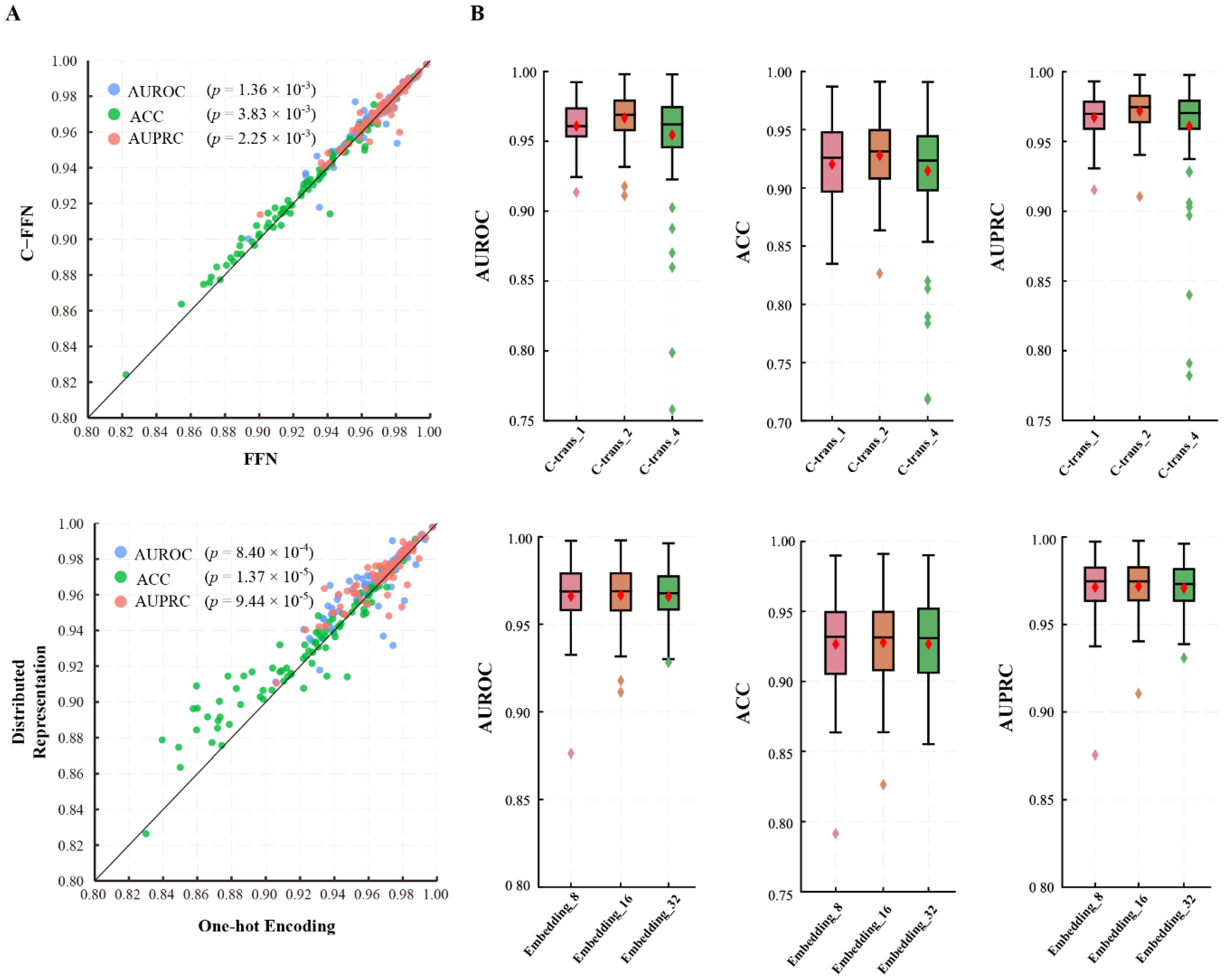
3.1.2. GHTNet Has the Optimal Structure
3.2. Effects on Different Chromatin Features on Tissue-Specific TF-DNA Binding
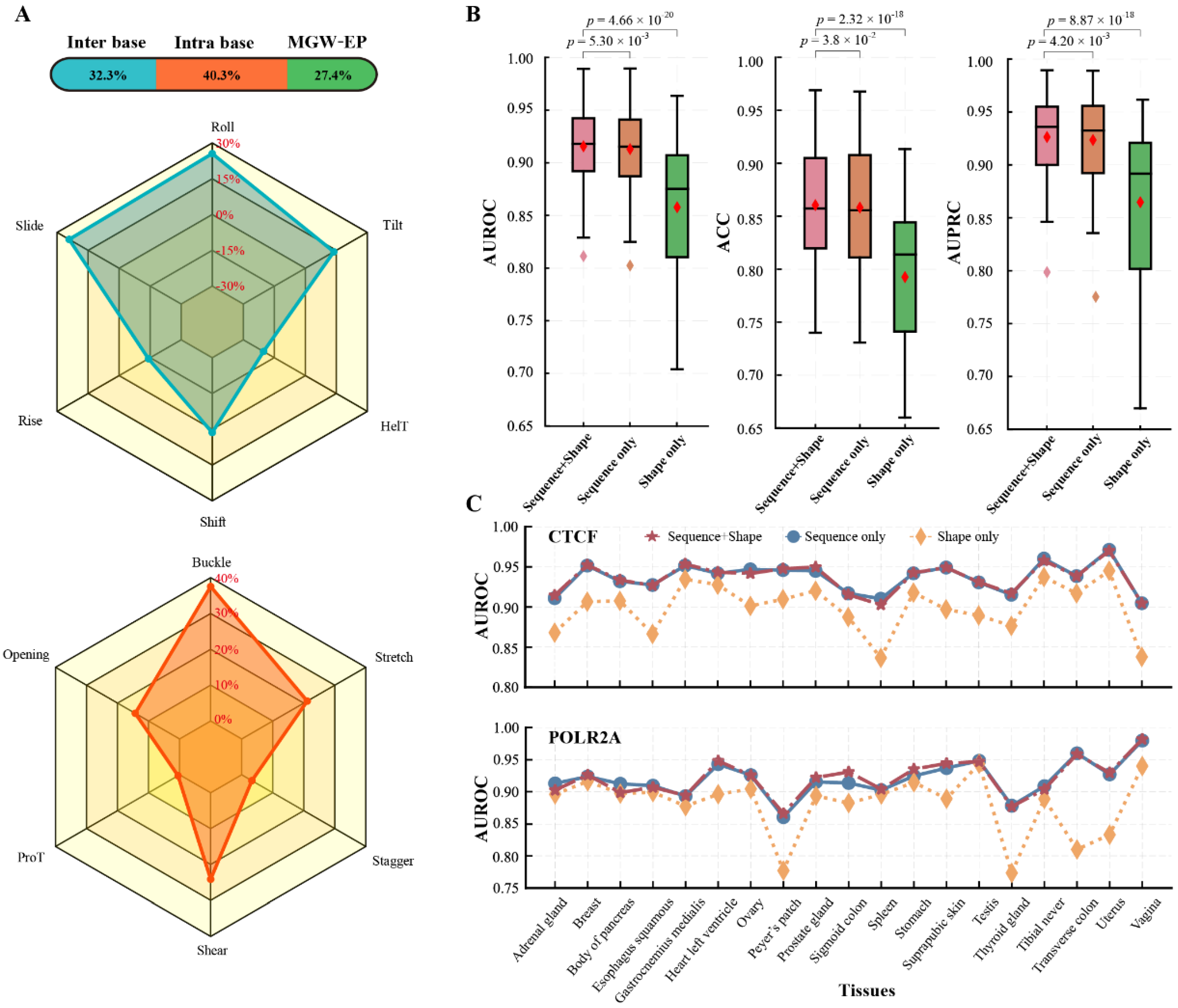
3.2.1. DNA Sequence and Shape Are Crucial Factors Affecting Tissue-Specific TF-DNA Binding
3.2.2. Combining Epigenomics Can Better Understand Tissue-Specific TF-DNA Binding
3.3. Mouse and Human TFs Exhibit Similarities in Tissue-Specific Binding
3.4. Downstream Applications
3.4.1. GHTNet Can Be Applied to Identify Important Gene Regions and Motifs
3.4.2. Significant Links between Transcription Factors, Chromatin Features, and Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Zhang, Q.; Hu, F.F.; Liu, C.J.; Guo, A.Y. A Comprehensive Survey for Human Transcription Factors on Expression, Regulation, Interaction, Phenotype and Cancer Survival. Brief. Bioinform. 2021, 22, bbab002. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D.; Mahony, S. Sequence and Chromatin Determinants of Transcription Factor Binding and the Establishment of Cell Type-Specific Binding Patterns. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2020, 1863, 194443. [Google Scholar] [CrossRef]
- Pal, S.; Hoinka, J.; Przytycka, T.M. Co-Select Reveals Sequence Non-Specific Contribution of DNA Shape to Transcription Factor Binding In Vitro. Nucleic Acids Res. 2019, 47, 6632–6641. [Google Scholar] [CrossRef] [Green Version]
- Vaishnav, E.D.; de Boer, C.G.; Molinet, J.; Yassour, M.; Fan, L.; Adiconis, X.; Thompson, D.A.; Levin, J.Z.; Cubillos, F.A.; Regev, A. The Evolution, Evolvability and Engineering of Gene Regulatory DNA. Nature 2022, 603, 455–463. [Google Scholar] [CrossRef]
- Phuycharoen, M.; Zarrineh, P.; Bridoux, L.; Amin, S.; Losa, M.; Chen, K.; Bobola, N.; Rattray, M. Uncovering Tissue-Specific Binding Features from Differential Deep Learning. Nucleic Acids Res. 2020, 48, e27. [Google Scholar] [CrossRef]
- Yan, J.; Qiu, Y.; Santos, A.M.R.D.; Yin, Y.; Li, Y.E.; Vinckier, N.; Nariai, N.; Benaglio, P.; Raman, A.; Li, X.; et al. Systematic Analysis of Binding of Transcription Factors to Noncoding Variants. Nature 2021, 591, 147–151. [Google Scholar] [CrossRef]
- De Almeida, B.P.; Reiter, F.; Pagani, M.; Stark, A. Deepstarr Predicts Enhancer Activity from DNA Sequence and Enables the De Novo Design of Synthetic Enhancers. Nat. Genet. 2022, 54, 613–624. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, A.; Zhao, J.; Xu, D.; Ma, Q.; Wang, Y. Assessing deep learning methods in cis-regulatory motif finding based on genomic sequencing data. Brief. Bioinform. 2022, 23, bbab374. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, Y.; Liu, Y.; Xiong, S.; Wang, M.; Zhou, J.; Zhang, Y. Towards a better understanding of TF-DNA binding prediction from genomic features. Comput. Biol. Med. 2022, 149, 105993. [Google Scholar] [CrossRef] [PubMed]
- Avsec, Ž.; Weilert, M.; Shrikumar, A.; Krueger, S.; Alexandari, A.; Dalal, K.; Fropf, R.; McAnany, C.; Gagneur, J.; Kundaje, A.; et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 2021, 53, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Park, C.Y.; Theesfeld, C.L.; Troyanskaya, O.G. An automated framework for efficiently designing deep convolutional neural networks in genomics. Nat. Mach. Intell. 2021, 3, 392–400. [Google Scholar] [CrossRef]
- Shen, L.-C.; Liu, Y.; Song, J.; Yu, D.-J. SAResNet: Self-attention residual network for predicting DNA-protein binding. Brief. Bioinform. 2021, 22, bbab101. [Google Scholar] [CrossRef]
- Quang, D.; Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; He, Y.; Chen, Z.; Zhang, Q. FCNGRU: Locating Transcription Factor Binding Sites by combing Fully Convolutional Neural Network with Gated Recurrent Unit. IEEE J. Biomed. Health Inform. 2021, 26, 1883–1890. [Google Scholar] [CrossRef]
- Wang, T.; Shao, W.; Huang, Z.; Tang, H.; Zhang, J.; Ding, Z.; Huang, K. MOGONET integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat. Commun. 2021, 12, 3445. [Google Scholar] [CrossRef]
- Li, J.; Sagendorf, J.M.; Chiu, T.-P.; Pasi, M.; Perez, A.; Rohs, R. Expanding the repertoire of DNA shape features for genome-scale studies of transcription factor binding. Nucleic Acids Res. 2017, 45, 12877–12887. [Google Scholar] [CrossRef]
- Xin, B.; Rohs, R. Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. 2018, 28, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Ma, A.; Hoppe, A.D.; Wang, C.; Li, Y.; Zhang, C.; Wang, Y.; Liu, B.; Ma, Q. Prediction of regulatory motifs from human Chip-sequencing data using a deep learning framework. Nucleic Acids Res. 2019, 47, 7809–7824. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Zeng, Y.; Liu, Y.; Xiong, S.; Wang, M.; Zhou, J.; Zou, Q. A novel convolution attention model for predicting transcription factor binding sites by combination of sequence and shape. Brief. Bioinform. 2022, 23, bbab525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Y.; Wang, S.; Chen, Z.-H.; Gribova, V.V.; Filaretov, V.F.; Huang, D.-S. Predicting in-vitro DNA protein binding with a spatially aligned fusion of sequence and shape. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Z.; Liu, Y.; Lu, L.; Tan, X.; Zou, Q. By hybrid neural networks for prediction and interpretation of transcription factor binding sites based on multi-omics. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021. [Google Scholar]
- Quang, D.; Xie, X. FactorNet: A deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods 2019, 166, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. Adv. Neural Inf. Process. Syst. 2017, 30, 5999–6009. [Google Scholar]
- Xu, P.; Zhu, X.; Clifton, D.A. Multimodal Learning with Transformers: A Survey. arXiv 2022, arXiv:2206.06488. [Google Scholar]
- Yang, M.; Huang, L.; Huang, H.; Tang, H.; Zhang, N.; Yang, H.; Wu, J.; Mu, F. Integrating Convolution and Self-Attention Improves Language Model of Human Genome for Interpreting Non-Coding Regions at Base-Resolution. Nucleic Acids Res. 2022, 50, e81. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with Alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ji, Y.; Zhou, Z.; Liu, H.; Davuluri, R.V. DNABERT: Pre-Trained Bidirectional Encoder Representations from Transformers Model for DNA-Language in Genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef]
- Ghandi, M.; Mohammad-Noori, M.; Ghareghani, N.; Lee, D.; Garraway, L.; Beer, M.A. gkmSVM: An R package for gapped-kmer SVM. Bioinformatics 2016, 32, 2205–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Zeng, H.; Edwards, M.D.; Liu, G.; Gifford, D.K. Convolutional neural network architectures for predicting DNA–protein binding. Bioinformatics 2016, 32, i121–i127. [Google Scholar] [CrossRef]
- Zhang, Q.; Shen, Z.; Huang, D.-S. Predicting in-vitro transcription factor binding sites using DNA sequence+ shape. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 667–676. [Google Scholar] [CrossRef]
- Gonzales, M.M.; Garbarino, V.R.; Pollet, E.; Palavicini, J.P.; Kellogg, D.L.; Kraig, E.; Orr, M.E. Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Investig. 2022, 132, e158453. [Google Scholar] [CrossRef]
- Lan, Z.; Chen, M.; Goodman, S.; Gimpel, K.; Sharma, P.; Soricut, R. Albert: A Lite Bert for Self-Supervised Learning of Language Representations. arXiv 2019, arXiv:1909.11942. [Google Scholar]
- Yang, Z.; Dai, Z.; Yang, Y.; Carbonell, J.; Salakhutdinov, R.R.; Le, Q.V. Xlnet: Generalized autoregressive pretraining for language understanding. Adv. Neural Inf. Process. Syst. 2019, 32, 5753–5763. [Google Scholar]
- Ullah, F.; Ben-Hur, A. A self-attention model for inferring cooperativity between regulatory features. Nucleic Acids Res. 2021, 49, e77. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ezer, D.; Adryan, B.; Stevens, T.J. Canonical and single-cell Hi-C reveal distinct chromatin interaction sub-networks of mammalian transcription factors. Genome Biol. 2018, 19, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belton, J.-M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanchantin, J.; Qi, Y. Graph convolutional networks for epigenetic state prediction using both sequence and 3D genome data. Bioinformatics 2020, 36, i659–i667. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protoc. 2010, 2, pdb.prot5384. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Hadsell, R.; LeCun, Y. Learning a similarity metric discriminatively, with application to face verification. In Proceedings of the 2005 IEEE Computer Society Conference on Computer Vision and Pattern Recognition (CVPR’05); San Diego, CA, USA: 20–25 June 2005. [CrossRef]
- Lecun, Y.; Bottou, L.; Bengio, Y.; Haffner, P. Gradient-based learning applied to document recognition. Proc. IEEE 1998, 86, 2278–2324. [Google Scholar] [CrossRef] [Green Version]
- Cybenko, G. Approximation by superpositions of a sigmoidal function. Math. Control Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. In Proceedings of the 25th International Conference on Neural Information Processing Systems, Lake Tahoe, NV, USA, 3–6 December 2012; Volume 1, pp. 1097–1105. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | AUROC | p-Value | Acc | p-Value | AUPRC | p-Value |
|---|---|---|---|---|---|---|
| GHTNet | 0.9667 | 0.9279 | 0.9718 | |||
| GHTNet-DNA | 0.9130 | 6.85 × 10−25 | 0.8582 | 2.34 × 10−24 | 0.9235 | 1.93 × 10−20 |
| DeepSEA | 0.9080 | 7.10 × 10−25 | 0.8450 | 2.04 × 10−27 | 0.9184 | 1.14 × 10−21 |
| CNN_Zeng | 0.9086 | 1.62 × 10−23 | 0.8477 | 1.30 × 10−25 | 0.9190 | 6.38 × 10−20 |
| DanQ | 0.9121 | 5.16 × 10−24 | 0.8487 | 3.80 × 10−25 | 0.9215 | 1.69 × 10−20 |
| DLBSS | 0.9079 | 2.20 × 10−23 | 0.8430 | 2.14 × 10−27 | 0.9164 | 5.78 × 10−20 |
| FactorNet | 0.9193 | 1.45 × 10−19 | 0.8641 | 1.55 × 10−19 | 0.9309 | 1.62 × 10−15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Liu, Y.; Wang, Z.; Wang, M.; Xiong, S.; Huang, G.; Gong, M. Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model. Genes 2022, 13, 1952. https://doi.org/10.3390/genes13111952
Zhang Y, Liu Y, Wang Z, Wang M, Xiong S, Huang G, Gong M. Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model. Genes. 2022; 13(11):1952. https://doi.org/10.3390/genes13111952
Chicago/Turabian StyleZhang, Yongqing, Yuhang Liu, Zixuan Wang, Maocheng Wang, Shuwen Xiong, Guo Huang, and Meiqin Gong. 2022. "Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model" Genes 13, no. 11: 1952. https://doi.org/10.3390/genes13111952
APA StyleZhang, Y., Liu, Y., Wang, Z., Wang, M., Xiong, S., Huang, G., & Gong, M. (2022). Uncovering the Relationship between Tissue-Specific TF-DNA Binding and Chromatin Features through a Transformer-Based Model. Genes, 13(11), 1952. https://doi.org/10.3390/genes13111952