
3D Genome Plasticity in Normal and Diseased Neurodevelopment


{kind=link}
{kind=link}
Abstract
:1. Introduction
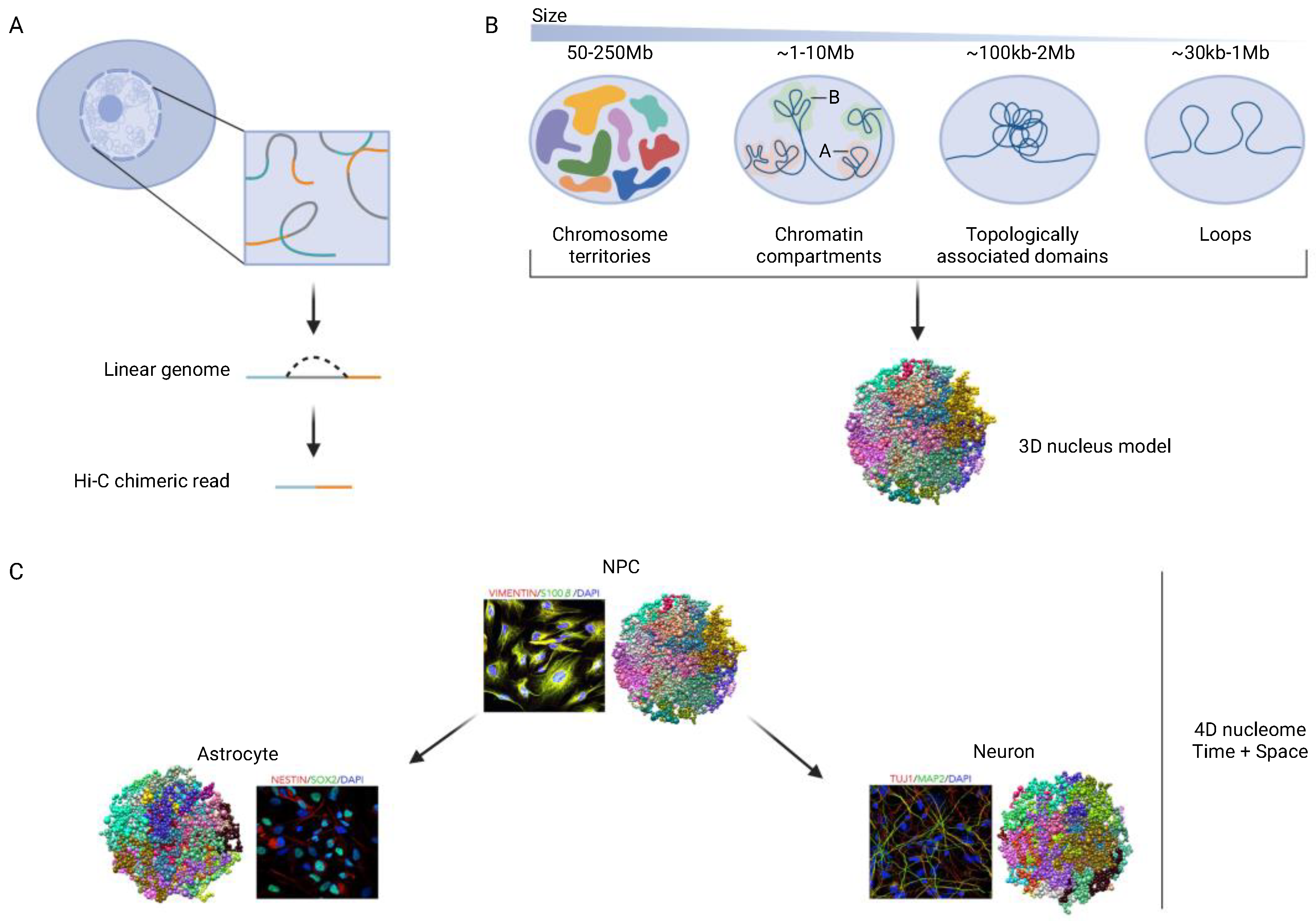
1.1. Spatial Chromosomal Organization and Neuroplasticity in Development
1.2. Spatial Genome Organization and Brain Evolution
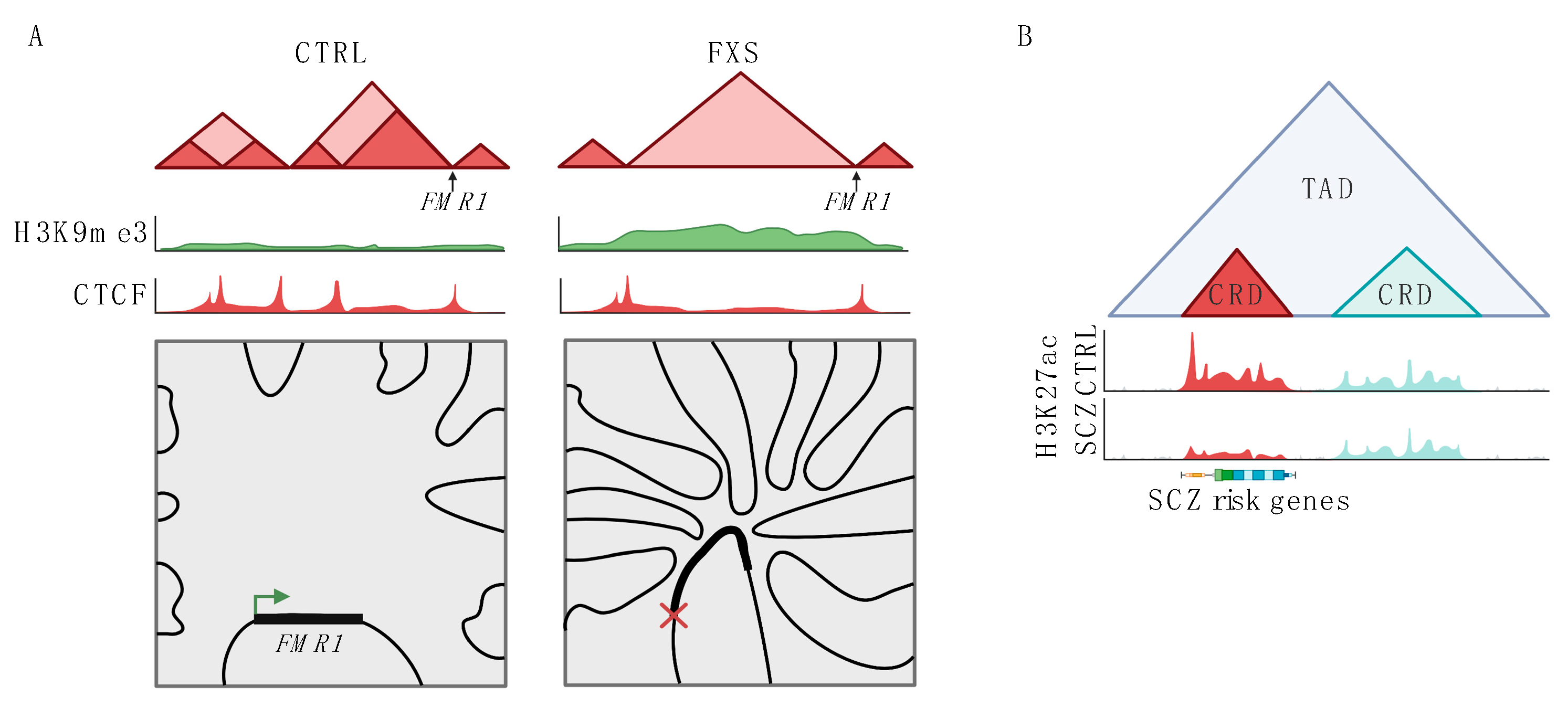
1.3. Neurological Disease Associated with Disrupted Organization of the 3D Genome
1.4. 3D Genome Organization in Context of Infection and Neuroinflammation
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hangauer, M.J.; Vaughn, I.W.; McManus, M.T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, e1003569. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef] [PubMed]
- BRAIN Iniatitive Cell Census Network. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 2021, 598, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Bryois, J.; Bakken, T.E.; Breen, G.; Crowley, J.J.; Gaspar, H.A.; Giusti-Rodriguez, P.; Hodge, R.D.; Miller, J.A.; Munoz-Manchado, A.B.; et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 2018, 50, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Trubetskoy, V.; Pardinas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508. [Google Scholar] [CrossRef]
- Ziffra, R.S.; Kim, C.N.; Ross, J.M.; Wilfert, A.; Turner, T.N.; Haeussler, M.; Casella, A.M.; Przytycki, P.F.; Keough, K.C.; Shin, D.; et al. Single-cell epigenomics reveals mechanisms of human cortical development. Nature 2021, 598, 205–213. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622.e1616. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, M.; Paulsen, J.; Jost, D.; Marti-Renom, M.A. 4D nucleome modeling. Curr. Opin. Genet. Dev. 2021, 67, 25–32. [Google Scholar] [CrossRef]
- Dekker, J.; Belmont, A.S.; Guttman, M.; Leshyk, V.O.; Lis, J.T.; Lomvardas, S.; Mirny, L.A.; O’Shea, C.C.; Park, P.J.; Ren, B.; et al. The 4D nucleome project. Nature 2017, 549, 219–226. [Google Scholar] [CrossRef]
- Espeso-Gil, S.; Halene, T.; Bendl, J.; Kassim, B.; Ben Hutta, G.; Iskhakova, M.; Shokrian, N.; Auluck, P.; Javidfar, B.; Rajarajan, P.; et al. A chromosomal connectome for psychiatric and metabolic risk variants in adult dopaminergic neurons. Genome Med. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, J.; Liyakat Ali, T.M.; Collas, P. Computational 3D genome modeling using Chrom3D. Nat. Protoc. 2018, 13, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.B.; Newman, A.G. On the relations of phase separation and Hi-C maps to epigenetics. R. Soc. Open Sci. 2020, 7, 191976. [Google Scholar] [CrossRef] [Green Version]
- Erdel, F.; Rippe, K. Formation of Chromatin Subcompartments by Phase Separation. Biophys J. 2018, 114, 2262–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eeftens, J.M.; Kapoor, M.; Michieletto, D.; Brangwynne, C.P. Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat. Commun. 2021, 12, 5888. [Google Scholar] [CrossRef] [PubMed]
- Belaghzal, H.; Borrman, T.; Stephens, A.D.; Lafontaine, D.L.; Venev, S.V.; Weng, Z.; Marko, J.F.; Dekker, J. Liquid chromatin Hi-C characterizes compartment-dependent chromatin interaction dynamics. Nat. Genet. 2021, 53, 367–378. [Google Scholar] [CrossRef]
- Nuebler, J.; Fudenberg, G.; Imakaev, M.; Abdennur, N.; Mirny, L.A. Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. USA 2018, 115, E6697–E6706. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, W.; Abdennur, N.; Goloborodko, A.; Pekowska, A.; Fudenberg, G.; Loe-Mie, Y.; Fonseca, N.A.; Huber, W.; Haering, C.H.; Mirny, L.; et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 2017, 551, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Won, H.; de la Torre-Ubieta, L.; Stein, J.L.; Parikshak, N.N.; Huang, J.; Opland, C.K.; Gandal, M.J.; Sutton, G.J.; Hormozdiari, F.; Lu, D.; et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 2016, 538, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572.e524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrie, J.L.; Lupo, M.S.; Xu, B.; Al Diri, I.; Valentine, M.; Putnam, D.; Griffiths, L.; Zhang, J.; Johnson, D.; Easton, J.; et al. Nucleome Dynamics during Retinal Development. Neuron 2019, 104, 512–528.e511. [Google Scholar] [CrossRef]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casino, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winick-Ng, W.; Kukalev, A.; Harabula, I.; Zea-Redondo, L.; Szabo, D.; Meijer, M.; Serebreni, L.; Zhang, Y.; Bianco, S.; Chiariello, A.M.; et al. Cell-type specialization is encoded by specific chromatin topologies. Nature 2021, 599, 684–691. [Google Scholar] [CrossRef]
- Zylka, M.J.; Simon, J.M.; Philpot, B.D. Gene length matters in neurons. Neuron 2015, 86, 353–355. [Google Scholar] [CrossRef] [Green Version]
- Calderon, L.; Weiss, F.D.; Beagan, J.A.; Oliveira, M.S.; Georgieva, R.; Wang, Y.F.; Carroll, T.S.; Dharmalingam, G.; Gong, W.; Tossell, K.; et al. Cohesin-dependence of neuronal gene expression relates to chromatin loop length. Elife 2022, 11, e76539. [Google Scholar] [CrossRef]
- Fernandez-Albert, J.; Lipinski, M.; Lopez-Cascales, M.T.; Rowley, M.J.; Martin-Gonzalez, A.M.; Del Blanco, B.; Corces, V.G.; Barco, A. Immediate and deferred epigenomic signatures of in vivo neuronal activation in mouse hippocampus. Nat. Neurosci. 2019, 22, 1718–1730. [Google Scholar] [CrossRef]
- Espeso-Gil, S.; Holik, A.Z.; Bonnin, S.; Jhanwar, S.; Chandrasekaran, S.; Pique-Regi, R.; Albaiges-Rafols, J.; Maher, M.; Permanyer, J.; Irimia, M.; et al. Environmental Enrichment Induces Epigenomic and Genome Organization Changes Relevant for Cognition. Front. Mol. Neurosci. 2021, 14, 664912. [Google Scholar] [CrossRef]
- Marco, A.; Meharena, H.S.; Dileep, V.; Raju, R.M.; Davila-Velderrain, J.; Zhang, A.L.; Adaikkan, C.; Young, J.Z.; Gao, F.; Kellis, M.; et al. Mapping the epigenomic and transcriptomic interplay during memory formation and recall in the hippocampal engram ensemble. Nat. Neurosci. 2020, 23, 1606–1617. [Google Scholar] [CrossRef]
- Rocks, D.; Shukla, M.; Ouldibbat, L.; Finnemann, S.C.; Kalluchi, A.; Rowley, M.J.; Kundakovic, M. Sex-specific multi-level 3D genome dynamics in the mouse brain. Nat. Commun. 2022, 13, 3438. [Google Scholar] [CrossRef] [PubMed]
- Vietri Rudan, M.; Barrington, C.; Henderson, S.; Ernst, C.; Odom, D.T.; Tanay, A.; Hadjur, S. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015, 10, 1297–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Liu, Y.; Dang, D.; Hu, T.; Hou, Y.; Meng, X.; Zhang, F.; Li, T.; Wang, C.; Li, M.; et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 2021, 184, 723–740.e721. [Google Scholar] [CrossRef] [PubMed]
- Shulha, H.P.; Crisci, J.L.; Reshetov, D.; Tushir, J.S.; Cheung, I.; Bharadwaj, R.; Chou, H.J.; Houston, I.B.; Peter, C.J.; Mitchell, A.C.; et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012, 10, e1001427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafin, R.N.; Khusnutdinova, E.K. Involvement of transposable elements in neurogenesis. Vavilovskii Zhurnal Genet. Sel. 2020, 24, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Grandi, N.; Tramontano, E.; Dieci, G. Retrotransposons as Drivers of Mammalian Brain Evolution. Life 2021, 11, 376. [Google Scholar] [CrossRef]
- Zhang, X.O.; Gingeras, T.R.; Weng, Z. Genome-wide analysis of polymerase III-transcribed Alu elements suggests cell-type-specific enhancer function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, S.; Espeso-Gil, S.; Loh, Y.E.; Javidfar, B.; Kassim, B.; Zhu, Y.; Zhang, Y.; Dong, Y.; Bicks, L.K.; Li, H.; et al. Neuron-specific chromosomal megadomain organization is adaptive to recent retrotransposon expansions. Nat. Commun. 2021, 12, 7243. [Google Scholar] [CrossRef]
- Nellaker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45. [Google Scholar] [CrossRef] [Green Version]
- Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. [Google Scholar] [CrossRef]
- Weiss, F.D.; Calderon, L.; Wang, Y.F.; Georgieva, R.; Guo, Y.; Cvetesic, N.; Kaur, M.; Dharmalingam, G.; Krantz, I.D.; Lenhard, B.; et al. Neuronal genes deregulated in Cornelia de Lange Syndrome respond to removal and re-expression of cohesin. Nat. Commun. 2021, 12, 2919. [Google Scholar] [CrossRef] [PubMed]
- Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G.; Santoro, S.L.; Shuss, C.; Ziegler, A.; Bonneau, D.; et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 2019, 21, 2723–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feurle, P.; Abentung, A.; Cera, I.; Wahl, N.; Ablinger, C.; Bucher, M.; Stefan, E.; Sprenger, S.; Teis, D.; Fischer, A.; et al. SATB2-LEMD2 interaction links nuclear shape plasticity to regulation of cognition-related genes. EMBO J. 2021, 40, e103701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Zhu, X.; Purmann, C.; Haney, M.S.; Ward, T.; Khechaduri, A.; Yao, J.; Weissman, S.M.; Urban, A.E. Local and global chromatin interactions are altered by large genomic deletions associated with human brain development. Nat. Commun. 2018, 9, 5356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, N.H.; Nevonen, K.A.; O’Connell, B.; McCann, C.; O’Neill, R.J.; Green, R.E.; Meyer, T.J.; Okhovat, M.; Carbone, L. Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res. 2018, 28, 983–997. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xue, B.; Zhang, M.; Zhang, L.; Hou, Y.; Qin, Y.; Long, H.; Su, Q.P.; Wang, Y.; Guan, X.; et al. Transcription-coupled structural dynamics of topologically associating domains regulate replication origin efficiency. Genome Biol. 2021, 22, 206. [Google Scholar] [CrossRef]
- Emerson, D.J.; Zhao, P.A.; Cook, A.L.; Barnett, R.J.; Klein, K.N.; Saulebekova, D.; Ge, C.; Zhou, L.; Simandi, Z.; Minsk, M.K.; et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature 2022, 606, 812–819. [Google Scholar] [CrossRef]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238.e215. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Zhou, L.; Ge, C.; Malachowski, T.; Kim, J.H.; Chandradoss, K.R.; Su, C.; Wu, H.; Rojas, A.; Wallace, O.; Titus, K.R.; et al. Spatially coordinated heterochromatinization of distal short tandem repeats in fragile X syndrome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Alcala-Vida, R.; Seguin, J.; Lotz, C.; Molitor, A.M.; Irastorza-Azcarate, I.; Awada, A.; Karasu, N.; Bombardier, A.; Cosquer, B.; Skarmeta, J.L.G.; et al. Age-related and disease locus-specific mechanisms contribute to early remodelling of chromatin structure in Huntington’s disease mice. Nat. Commun. 2021, 12, 364. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthoj, B. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network; Pathway Analysis Subgroup of the Psychiatric Genomics, Consortium Corrigendum: Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015, 18, 1861. [CrossRef] [PubMed]
- Takata, A.; Xu, B.; Ionita-Laza, I.; Roos, J.L.; Gogos, J.A.; Karayiorgou, M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron 2014, 82, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.J.; et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef]
- Roussos, P.; Mitchell, A.C.; Voloudakis, G.; Fullard, J.F.; Pothula, V.M.; Tsang, J.; Stahl, E.A.; Georgakopoulos, A.; Ruderfer, D.M.; Charney, A.; et al. A role for noncoding variation in schizophrenia. Cell Rep. 2014, 9, 1417–1429. [Google Scholar] [CrossRef]
- Hu, B.; Won, H.; Mah, W.; Park, R.B.; Kassim, B.; Spiess, K.; Kozlenkov, A.; Crowley, C.A.; Pochareddy, S.; Psych, E.C.; et al. Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders. Nat. Commun. 2021, 12, 3968. [Google Scholar] [CrossRef]
- Sey, N.Y.A.; Hu, B.; Mah, W.; Fauni, H.; McAfee, J.C.; Rajarajan, P.; Brennand, K.J.; Akbarian, S.; Won, H. A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 2020, 23, 583–593. [Google Scholar] [CrossRef]
- Powell, S.K.; O’Shea, C.; Townsley, K.; Prytkova, I.; Dobrindt, K.; Elahi, R.; Iskhakova, M.; Lambert, T.; Valada, A.; Liao, W.; et al. Induction of dopaminergic neurons for neuronal subtype-specific modeling of psychiatric disease risk. Mol. Psychiatry 2021, 1–13. [Google Scholar] [CrossRef]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.C.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- Ahanger, S.H.; Delgado, R.N.; Gil, E.; Cole, M.A.; Zhao, J.; Hong, S.J.; Kriegstein, A.R.; Nowakowski, T.J.; Pollen, A.A.; Lim, D.A. Distinct nuclear compartment-associated genome architecture in the developing mammalian brain. Nat. Neurosci. 2021, 24, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girdhar, K.; Hoffman, G.E.; Bendl, J.; Rahman, S.; Dong, P.; Liao, W.; Hauberg, M.E.; Sloofman, L.; Brown, L.; Devillers, O.; et al. Chromatin domain alterations linked to 3D genome organization in a large cohort of schizophrenia and bipolar disorder brains. Nat. Neurosci. 2022, 25, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lei, E.P. Function and regulation of chromatin insulators in dynamic genome organization. Curr. Opin. Cell Biol. 2019, 58, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Gao, Y.; Deep-Soboslay, A.; Tao, R.; Hyde, T.M.; Weinberger, D.R.; Kleinman, J.E. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 2016, 19, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Zazhytska, M.; Kodra, A.; Hoagland, D.A.; Frere, J.; Fullard, J.F.; Shayya, H.; McArthur, N.G.; Moeller, R.; Uhl, S.; Omer, A.D.; et al. Non-cell-autonomous disruption of nuclear architecture as a potential cause of COVID-19-induced anosmia. Cell 2022, 185, 1052–1064.e1012. [Google Scholar] [CrossRef]
- Luers, J.C.; Rokohl, A.C.; Loreck, N.; Wawer Matos, P.A.; Augustin, M.; Dewald, F.; Klein, F.; Lehmann, C.; Heindl, L.M. Olfactory and Gustatory Dysfunction in Coronavirus Disease 2019 (COVID-19). Clin. Infect. Dis. 2020, 71, 2262–2264. [Google Scholar] [CrossRef]
- Lomvardas, S.; Barnea, G.; Pisapia, D.J.; Mendelsohn, M.; Kirkland, J.; Axel, R. Interchromosomal interactions and olfactory receptor choice. Cell 2006, 126, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Clowney, E.J.; LeGros, M.A.; Mosley, C.P.; Clowney, F.G.; Markenskoff-Papadimitriou, E.C.; Myllys, M.; Barnea, G.; Larabell, C.A.; Lomvardas, S. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell 2012, 151, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, C.; Arendt, G.; Hahn, K.; Husstedt, I.W.; Maschke, M.; Neuen-Jacob, E.; Obermann, M.; Rosenkranz, T.; Schielke, E.; Straube, E. HIV-1-associated neurocognitive disorder: Epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 2017, 264, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza-Jennings, A.L.; Valada, A.; O’Shea, C.; Iskhakova, M.; Hu, B.; Javidfar, B.; Hutta, G.B.; Lambert, T.; Murray, J.; Kassim, B.; et al. HIV integration in the human brain is linked to microglial activation and 3D genome remodeling. bioRxiv 2022. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaza-Jennings, A.; Valada, A.; Akbarian, S. 3D Genome Plasticity in Normal and Diseased Neurodevelopment. Genes 2022, 13, 1999. https://doi.org/10.3390/genes13111999
Plaza-Jennings A, Valada A, Akbarian S. 3D Genome Plasticity in Normal and Diseased Neurodevelopment. Genes. 2022; 13(11):1999. https://doi.org/10.3390/genes13111999
Chicago/Turabian StylePlaza-Jennings, Amara, Aditi Valada, and Schahram Akbarian. 2022. "3D Genome Plasticity in Normal and Diseased Neurodevelopment" Genes 13, no. 11: 1999. https://doi.org/10.3390/genes13111999
APA StylePlaza-Jennings, A., Valada, A., & Akbarian, S. (2022). 3D Genome Plasticity in Normal and Diseased Neurodevelopment. Genes, 13(11), 1999. https://doi.org/10.3390/genes13111999