

Ehlers-Danlos: A Literature Review and Case Report in a Colombian Woman with Multiple Comorbidities

 , ,
, ,  , and
, and
Abstract
:1. Introduction
1.1. Clinical Manifestations and Pathogenesis
1.2. Classification
1.3. Management, Prognosis, and Complications
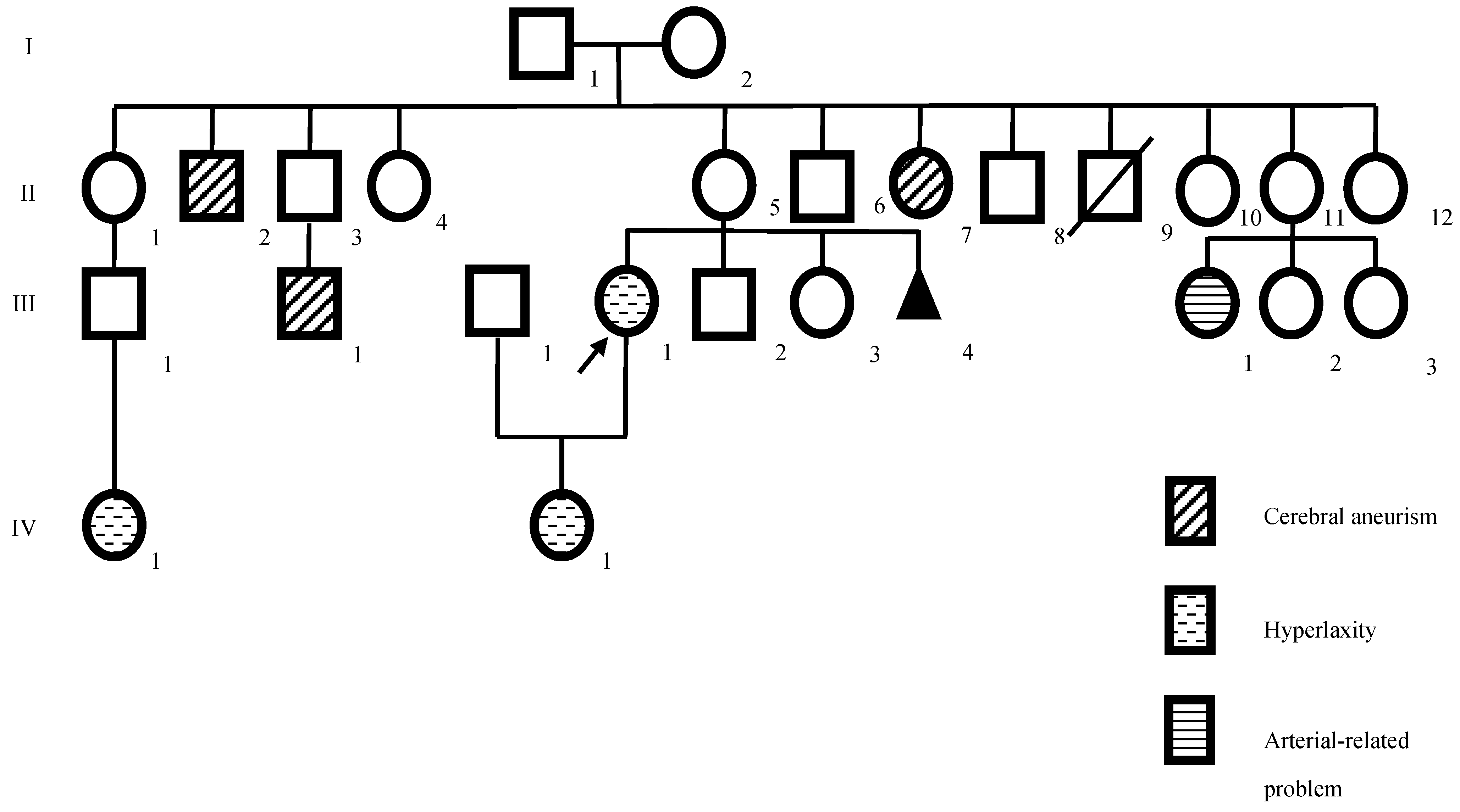
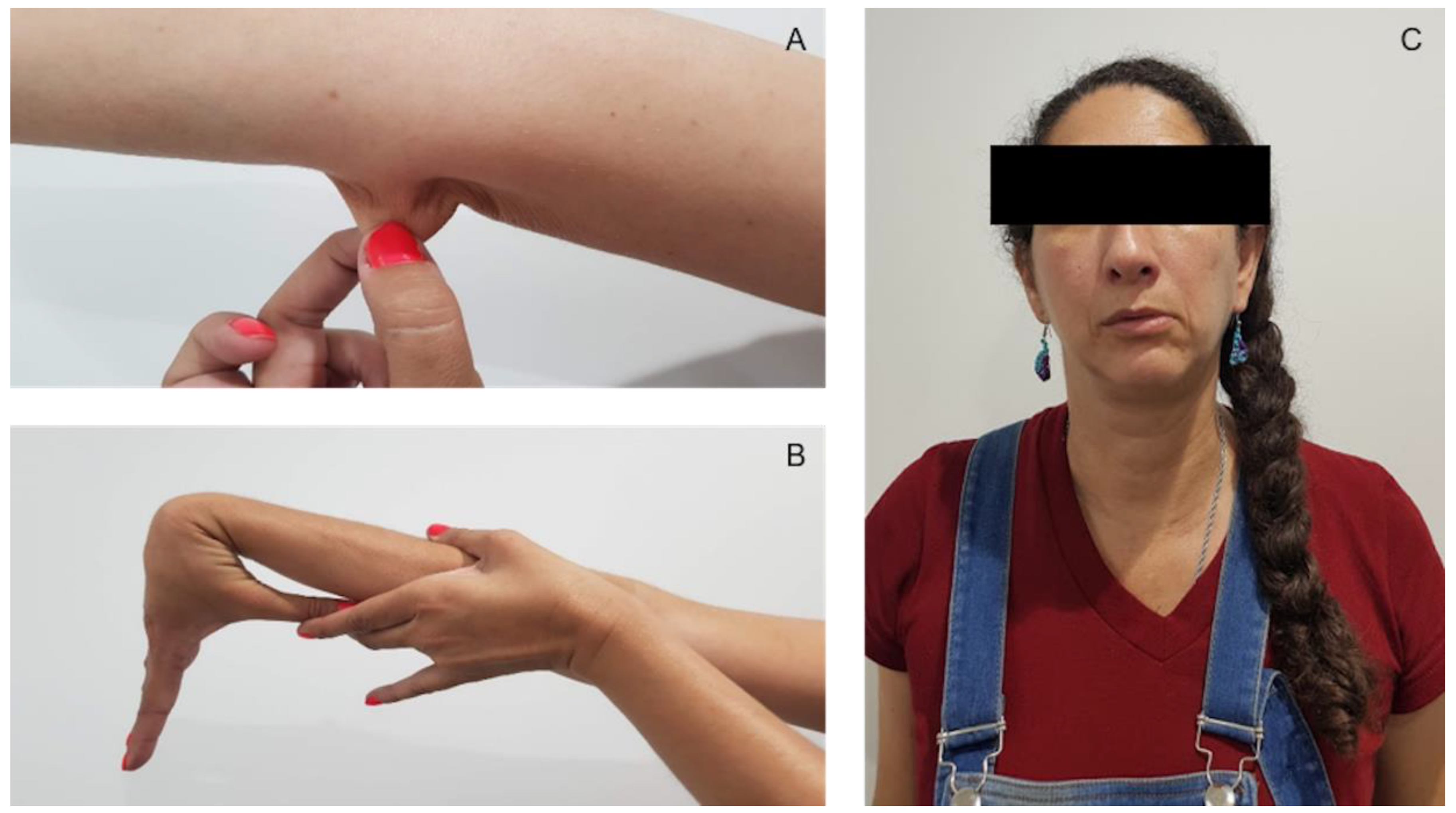
2. Case Presentation
2.1. Neurological and Psychological Assessment
2.2. Neurological Examination
2.3. Genetic Study
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yeowell, H.N.; Pinnell, S.R. The Ehlers-Danlos syndromes. Semin. Dermatol. 1993, 12, 229–240. [Google Scholar] [PubMed]
- Callewaert, B.; Malfait, F.; Loeys, B.; De Paepe, A. Ehlers-Danlos syndromes and Marfan syndrome. Best Pract. Res. Clin. Rheumatol. 2008, 22, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers–Danlos syndrome (a.k.a. Ehlers–Danlos syndrome Type III and Ehlers–Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- De Paepe, A.; Malfait, F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin. Genet. 2012, 82, 1–11. [Google Scholar] [CrossRef]
- Henderson, F.C.; Austin, C.; Benzel, E.; Bolognese, P.; Ellenbogen, R.; Francomano, C.A.; Ireton, C.; Klinge, P.; Koby, M.; Long, D.; et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes; Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 195–211. [Google Scholar] [CrossRef] [Green Version]
- Bulbena, A.; Pailhez, G.; Bulbena-Cabré, A.; Mallorquí-Bagué, N.; Baeza-Velasco, C. Joint hypermobility, anxiety and psychosomatics: Two and a half decades of progress toward a new phenotype. Adv. Psychosom. Med. 2015, 34, 143–157. [Google Scholar]
- Pasquini, M.; Celletti, C.; Berardelli, I.; Roselli, V.; Mastroeni, S.; Castori, M.; Biondi, M.; Camerota, F. Unexpected association between joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type and obsessive-compulsive personality disorder. Rheumatol. Int. 2014, 34, 631–636. [Google Scholar] [CrossRef]
- Voermans, N.C.; Knoop, H.; van de Kamp, N.; Hamel, B.C.; Bleijenberg, G.; van Engelen, B.G. Fatigue Is a Frequent and Clinically Relevant Problem in Ehlers-Danlos Syndrome. Semin. Arthritis Rheum. 2010, 40, 267–274. [Google Scholar] [CrossRef]
- Baeza-Velasco, C.; Bourdon, C.; Polanco-Carrasco, R.; De Jouvencel, M.; Gely-Nargeot, M.-C.; Gompel, A.; Hamonet, C. Cognitive impairment in women with joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. Rheumatol. Int. 2017, 37, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Malfait, F.; Renard, M.; André, J.; Hausser, I.; Loeys, B.; Coucke, P.; De Paepe, A. COL5A1 signal peptide mutations interfere with protein secretion and cause classic Ehlers-Danlos syndrome. Hum. Mutat. 2008, 30, E395–E403. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Coucke, P.; Symoens, S.; Loeys, B.; Nuytinck, L.; De Paepe, A. The molecular basis of classic Ehlers-Danlos syndrome: A comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 2004, 25, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Viglio, S.; Zoppi, N.; Sangalli, A.; Gallanti, A.; Barlati, S.; Mottes, M.; Colombi, M.; Valli, M. Rescue of migratory defects of Ehlers-Danlos syndrome fibroblasts in vitro by type V collagen but not insulin-like binding protein-1. J. Investig. Dermatol. 2008, 128, 1915–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birk, D.E. Type V collagen: Heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 2000, 32, 223–237. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A Recessive Form of the Ehlers–Danlos Syndrome Caused by Tenascin-X Deficiency. N. Engl. J. Med. 2002, 345, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Malfait, F.; Symoens, S.; Coucke, P.; Nunes, L.; De Almeida, S.; De Paepe, A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. J. Med. Genet. 2005, 43, e36. [Google Scholar] [CrossRef]
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722. [Google Scholar] [CrossRef]
- Yeowell, H.; Steinmann, B. PLOD1-Related Kyphoscoliotic Ehlers-Danlos Syndrome. In GeneReviews; Adam, M.P., Everman, D.B., Mirzaa, G.M., Eds.; University of Washington: Seattle, WA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1462/ (accessed on 12 September 2022).
- Giunta, C.; Superti-Furga, A.; Spranger, S.; Cole, W.G.; Steinmann, B. Ehlers-Danlos syndrome type VII: Clinical features and molecular defects. JBJS 1999, 81, 225–238. [Google Scholar] [CrossRef]
- Byers, P.H.; Duvic, M.; Atkinson, M.; Robinow, M.; Smith, L.T.; Krane, S.M.; Greally, M.T.; Ludman, M.; Matalon, R.; Pauker, S.; et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am. J. Med. Genet. 1997, 72, 94–105. [Google Scholar] [CrossRef]
- Colige, A.; Sieron, A.L.; Li, S.-W.; Schwarze, U.; Petty, E.; Wertelecki, W.; Wilcox, W.; Krakow, D.; Cohn, D.H.; Reardon, W.; et al. Human Ehlers-Danlos Syndrome Type VII C and Bovine Dermatosparaxis Are Caused by Mutations in the Procollagen I N-Proteinase Gene. Am. J. Hum. Genet. 1999, 65, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; De Paepe, A. The Ehlers-Danlos Syndrome. In Progress in Heritable Soft Connective Tissue Diseases; Advances in Experimental Medicine and Biology; Halper, J., Ed.; Springer: Dordrecht, The Netherlands, 2014; Volume 802. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Schwarze, U.; Jennings, J.F.; Byers, P.H. Molecular mechanisms of classical Ehlers-Danlos syndrome (EDS). Hum. Mutat. 2009, 30, 995–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Juul-Kristensen, B.; Rogind, H.; Jensen, D.V.; Remvig, L. Inter-examiner reproducibility of tests and criteria for generalized joint hypermobility and benign joint hypermobility syndrome. Rheumatology 2007, 46, 1835–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweers, M.C.; Dean, W.B.; van Kuppevelt, T.H.; Bristow, J.; Schalkwijk, J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin. Genet. 2005, 67, 330–334. [Google Scholar] [CrossRef]
- Danielson, K.G.; Baribault, H.; Holmes, D.F.; Graham, H.; Kadler, K.E.; Iozzo, R.V. Targeted disruption of decorin leads to abnormal collagen fibril morphology and skin fragility. J. Cell Biol. 1997, 136, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Svensson, L.; Aszódi, A.; Reinholt, F.P.; Fässler, R.; Heinegård, D.; Oldberg, Å. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J. Biol. Chem. 1999, 274, 9636–9647. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, S.; Magnuson, T.; Lass, J.H.; Jepsen, K.J.; LaMantia, C.; Carroll, H. Lumican regulates collagen fibril assembly: Skin fragility and corneal opacity in the absence of lumican. J. Cell Biol. 1998, 141, 1277–1286. [Google Scholar] [CrossRef]
- Mao, J.R.; Bristow, J. The Ehlers-Danlos syndrome: On beyond collagens. J. Clin. Investig. 2001, 107, 1063–1069. [Google Scholar] [CrossRef] [Green Version]
- Hamonet, C.; Brissot, R.; Gompel, A.; Baeza-Velasco, C.; Guinchat, V.; Brock, I.; Ducret, L.; Pommeret, S.; Metlaine, A. Prospective Study of 853 Patients. EC Neurol. 2018, 10, 428–439. [Google Scholar]
- Sulli, A.; Talarico, R.; Scirè, C.A.; Avcin, T.; Castori, M.; Ferraris, A.; Frank, C.; Grunert, J.; Paolino, S.; Bombardieri, S.; et al. Ehlers-Danlos syndromes: State of the art on clinical practice guidelines. RMD Open 2018, 4, e000790. [Google Scholar] [CrossRef] [PubMed]
- Baeza-Velasco, C.; Gély-Nargeot, M.C.; Vilarrasa, A.B.; Bravo, J.F. Joint hypermobility syndrome: Problems that require psychological intervention. Rheumatol. Int. 2011, 31, 1131–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Strand, M. Ehlers–Danlos syndrome in a young woman with anorexia nervosa and complex somatic symptoms. Int. J. Eat. Disord. 2017, 51, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Rewari, A.; Shanthanna, H. Management of chronic pain in Ehlers-Danlos syndrome. Medicine 2018, 97, e13115. [Google Scholar] [CrossRef] [PubMed]
- Sacheti, A.; Szemere, J.; Bernstein, B.; Tafas, T.; Schechter, N.; Tsipouras, P. Chronic pain is a manifestation of the Ehlers-Danlos syndrome. J. Pain Symptom Manag. 1997, 14, 88–93. [Google Scholar] [CrossRef]
- Voermans, N.C.; Knoop, H.; Bleijenberg, G.; Van Engelen, B.G. Pain in Ehlers-Danlos Syndrome is common, severe, and associated with functional impairment. J. Pain Symptom Manag. 2010, 40, 370–378. [Google Scholar] [CrossRef]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers–Danlos syndromes, rare types. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 70–115. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Okamoto, N.; Miyake, N.; Taira, K.; Sato, Y.; Matsuda, K.; Akimaru, N.; Ohashi, H.; Wakui, K.; Fukushima, Y.; et al. Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: Observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. Part A 2011, 155, 1949–1958. [Google Scholar] [CrossRef]
- Kosho, T. CHST14/D4ST1 deficiency: New form of Ehlers-Danlos syndrome. Pediatr. Int. 2016, 58, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Castori, M.; Morlino, S.; Celletti, C.; Ghibellini, G.; Bruschini, M.; Grammatico, P.; Blundo, C.; Camerota, F. Re-writing the natural history of pain and related symptoms in the joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am. J. Med. Genet. Part A 2013, 161, 2989–3004. [Google Scholar] [CrossRef]
- Chopra, P.; Tinkle, B.; Hamonet, C.; Brock, I.; Gompel, A.; Bulbena, A.; Francomano, C. Pain management in the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 212–219. [Google Scholar] [CrossRef]
- Hamonet, C.; Gompel, A.; Raffray, Y.; Zeitoun, J.D.; Delarue, M.; Vlamynck, E.; Haidari, R.; Mazaltarinej, G. Multiple pains in Ehlers-Danlos Syndrome. Description and proposal of a therapy protocol. Douleurs 2014, 15, 264–277. [Google Scholar]
- Hamonet, C.; Brock, I. Joint mobility and Ehlers-Danlos syndrome, (EDS) new data based on 232 cases. J. Arthritis 2015, 4, 1–5. [Google Scholar]
- Arthur, K.; Caldwell, K.; Forehand, S.; Davis, K. Pain control methods in use and perceived effectiveness by patients with Ehlers-Danlos syndrome: A descriptive study. Disabil. Rehabil. 2016, 38, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Ostrosky, F.; Gómez, M.E.; Matute, E.; Rosselli, M.; Ardila, A.P.D. Neuropsi: Atención y Memoria, 3rd ed.; Manual Moderno: Ciudad de México, México, 2019. [Google Scholar]
- Beck, A.T.; Steer, R.A. Beck Anxiety Inventory: Manual; Psychological Corporation: San Antonio, TX, USA, 1993; 23p. [Google Scholar]
- Beck, A.T.; Steer, R.A.; Brown, G.K. Beck Depression Inventory Manual; Psychological Corporation: San Antonio, TX, USA, 1993; 38p. [Google Scholar]
- Fernández, J.; Mielgo, L. Escalas de Apreciación del Estrés; Manual; TEA: Madrid, Spain, 2001. [Google Scholar]
- Costa, P.T.; McCrae, R.R. The NEO-PI/NEO-FFI Manual Supplement; Psychological Assessment Resources: Odessa, FL, USA, 1989. [Google Scholar]
- Mason, J.W.; Wang, S.; Yehuda, R.; Riney, S.; Charney, D.S.; Southwick, S.M. Psychogenic lowering of urinary cortisol levels linked to increased emotional numbing and a shame-depressive syndrome in combat-related posttraumatic stress disorder. Psychosom. Med. 2001, 63, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Vaser, R.; Adusumalli, S.; Leng, S.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Kole, A.; Faurisson, F. Rare diseases social epidemiology: Analysis of inequalities. Adv. Exp. Med. Biol. 2010, 686, 223–250. [Google Scholar]
- Tinkle, B.T.; Bird, H.A.; Grahame, R.; Lavallee, M.; Levy, H.P.; Sillence, D. The lack of clinical distinction between the hypermobility type of Ehlers-Danlos syndrome and the joint hypermobility syndrome (a.k.a. hypermobility syndrome). Am. J. Med. Genet. Part A 2009, 149, 2368–2370. [Google Scholar] [CrossRef]
- Bregant, T.; Spevak, M.K. Ehlers-Danlos Syndrome: Not Just Joint Hypermobility. Case Rep. Med. 2018, 2018, 5053825. [Google Scholar] [CrossRef] [Green Version]
- Barabas, A.P. Ehlers-Danlos Syndrome: Associated with Prematurity and Premature Rupture of Foetal Membranes; Possible Increase in Incidence. BMJ 1966, 2, 682–684. [Google Scholar] [CrossRef] [Green Version]
- Bravo, J. Ehlers-Danlos syndrome, with special emphasis in the joint hypermobility syndrome. Rev. Med. Chil. 2009, 137, 1488–1497. [Google Scholar]
- Scheper, M.C.; Nicholson, L.L.; Adams, R.D.; Tofts, L.; Pacey, V. The natural history of children with joint hypermobility syndrome and Ehlers-Danlos hypermobility type: A longitudinal cohort study. Rheumatology 2017, 56, 2073–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, T.G.; Lichtenstein, J.R.; Moore, T.L.; Weiss, T.; Zuckner, J. Ehlers-Danlos syndrome presenting as rheumatic manifestations in the child. J. Rheumatol. 1981, 8, 79–85. [Google Scholar] [PubMed]
- Castori, M.; Morlino, S.; Pascolini, G.; Blundo, C.; Grammatico, P. Gastrointestinal and nutritional issues in joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am. J. Med. Genet. Part C Semin. Med. Genet. 2015, 169, 54–75. [Google Scholar] [CrossRef] [PubMed]
- Fikree, A.; Chelimsky, G.; Collins, H.; Kovacic, K.; Aziz, Q. Gastrointestinal involvement in the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarate, N.; Farmer, A.D.; Grahame, R.; Mohammed, S.D.; Knowles, C.H.; Scott, S.M.; Aziz, Q. Unexplained gastrointestinal symptoms and joint hypermobility: Is connective tissue the missing link? Neurogastroenterol. Motil. 2010, 22, 252-e78. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, A.; Malfait, F. Bleeding and bruising in patients with Ehlers-Danlos syndrome and other collagen vascular disorders. Br. J. Haematol. 2004, 127, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Haviarova, Z.; Janega, P.; Durdik, S.; Kovac, P.; Mraz, P.; Stvrtinova, V. Comparison of collagen subtype I and III presence in varicose and non-varicose vein walls. Bratisl. Lek. Listy 2008, 109, 102–105. [Google Scholar]
- Sansilvestri-Morel, P.; Rupin, A.; Badier-Commander, C.; Kern, P.; Fabiani, J.-N.; Verbeuren, T.J.; Vanhoutte, P.M. Imbalance in the synthesis of collagen type I and collagen type III in smooth muscle cells derived from human varicose veins. J. Vasc. Res. 2001, 38, 560–568. [Google Scholar] [CrossRef]
- Lim, C.S.; Davies, A.H. Pathogenesis of primary varicose veins. Br. J. Surg. 2009, 96, 1231–1242. [Google Scholar] [CrossRef]
- Tinkle, B.T. Joint Hypermobility Handbook: A Guide for the Issues & Management of Ehlers-Danlos Syndrome Hypermobility Type and the Hypermobility Syndrome; Left Paw Press: Greens Fork, IN, USA, 2010; 251p. [Google Scholar]
- Lammers, K.; Lince, S.L.; Spath, M.A.; van Kempen, L.C.; Hendriks, J.; Vierhout, M.E.; Kluivers, K.B. Pelvic organ prolapse and collagen-associated disorders. Int. Urogynecol. J. 2012, 23, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Ericson, W.B.; Wolman, R. Orthopaedic management of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- al-Rawi, Z.; Nessan, A.H. Joint hypermobility in patients with chondromalacia patellae. Br. J. Rheumatol. 1997, 36, 1324–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehan, F.T.; Derasari, A.; Brindle, T.J.; Alter, K.E. Understanding patellofemoral pain with maltracking in the presence of joint laxity: Complete 3D in vivo patellofemoral and tibiofemoral kinematics. J. Orthop. Res. 2008, 27, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoler, J.M.; Oaklander, A.L. Patients with Ehlers Danlos syndrome and CRPS: A possible association? Pain 2006, 123, 204–209. [Google Scholar] [CrossRef]
- Weir, F.W.; Hatch, J.L.; Muus, J.S.; Wallace, S.A.; Meyer, T.A. Audiologic Outcomes in Ehlers-Danlos Syndrome. Otol. Neurotol. 2016, 37, 748–752. [Google Scholar] [CrossRef]
- Chau, A.S.; Jongco, A.M. Allergic and Immunologic Dysregulation in Ehlers-Danlos Syndrome: A Case Series. J. Allergy Clin. Immunol. 2018, 141, AB125. [Google Scholar] [CrossRef]
- Smith, T.O.; Easton, V.; Bacon, H.; Jerman, E.; Armon, K.; Poland, F.; Macgregor, A.J. The relationship between benign joint hypermobility syndrome and psychological distress: A systematic review and meta-analysis. Rheumatology 2013, 53, 114–122. [Google Scholar] [CrossRef]
- Rubio-Agusti, I.; Kojovic, M.; Chandrashekar, H.S.; Edwards, M.J.; Bhatia, K.P. Cervical dystonia and joint hypermobility syndrome: A dangerous combination. Mov. Disord. 2012, 27, 203–204. [Google Scholar] [CrossRef]
- Walter, S. Case Report: Ehlers-Danlos syndrome in an adolescent presenting with Chronic Daily Headache. Surg. Neurol. Int. 2014, 5, 475–478. [Google Scholar] [CrossRef]
- Puledda, F.; Vigano, A.; Celletti, C.; Petolicchio, B.; Toscano, M.; Vicenzini, E.; Castori, M.; Laudani, G.; Valente, D.; Camerota, F.; et al. A study of migraine characteristics in joint hypermobility syndrome a.k.a. Ehlers-Danlos syndrome, hypermobility type. Neurol. Sci. 2015, 36, 1417–1424. [Google Scholar] [CrossRef]
- Mitra, A.; Ramakrishnan, R.; Kader, M.A. Open angle glaucoma in a case of Type IV Ehler Danlos syndrome: A rarely reported association. Indian J. Ophthalmol. 2014, 62, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; Gui, J.; Dinulos, M.B.P.; Chou, R.C. Ehlers-Danlos syndrome hypermobility type is associated with rheumatic diseases. Sci. Rep. 2017, 7, 39636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dousa, K.M.; Khan, K.; Alencherry, B.; Deng, L.; Salata, R.A. Renal infarction in vascular Ehlers–Danlos syndrome masquerading as pyelonephritis. Clin. Case Rep. 2018, 6, 1478–1480. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.J.; Mallett, V.T.; Frahm, J.D.; A Richardson, D.; Evans, M.I. Gynecologic disorders in women with Ehlers-Danlos syndrome. J. Soc. Gynecol. Investig. 1995, 2, 559–564. [Google Scholar] [CrossRef]
- Brunk, I.; Ikonomidou, C.; Neumann, L.M. Ehlers-Danlos syndrome type VI with cystic malformations of the meninges in a 7-year-old girl. Eur. J. Pediatr. 2004, 163, 214–217. [Google Scholar] [CrossRef]
- Grosveld, W.J.H.M.; Gilhuis, H.; Voermans, N. Spontaneous intracranial hypotension in a patient with classical type Ehlers-Danlos syndrome. Neurol. India 2011, 59, 633–634. [Google Scholar]
- Castori, M.; Morlino, S.; Celletti, C.; Celli, M.; Morrone, A.; Colombi, M.; Camerota, F.; Grammatico, P. Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome, hypermobility type): Principles and proposal for a multidisciplinary approach. Am. J. Med. Genet. A 2012, 158, 2055–2270. [Google Scholar] [CrossRef]
- Pavan, S.; Rommel, K.; Mateo-Marquina, M.E.; Höhn, S.; Lanneau, V.; Rath, A. Clinical Practice Guidelines for Rare Diseases: The Orphanet Database. PLoS ONE 2017, 12, e0170365. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Spatz, M.A. Genetics Home Reference. J. Med. Libr. Assoc. 2004, 92, 282–283. [Google Scholar]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zwolanek, D.; Izu, Y.; Gandhy, S.; Schreiber, G.; Brockmann, K.; Devoto, M.; Tian, Z.; Hu, Y.; Veit, G.; et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum. Mol. Genet. 2013, 23, 2339–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punetha, J.; Kesari, A.; Hoffman, E.P.; Gos, M.; Kamińska, A.; Kostera-Pruszczyk, A.; Hausmanowa-Petrusewicz, I.; Hu, Y.; Zou, Y.; Bönnemann, C.G.; et al. Novel Col12A1 variant expands the clinical picture of congenital myopathies with extracellular matrix defects. Muscle Nerve 2017, 55, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbaere, S.; Dhooge, T.; Syx, D.; Petit, F.; Goemans, N.; Destrée, A.; Vanakker, O.; De Rycke, R.; Symoens, S.; Malfait, F. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet. Med. 2020, 22, 112–123. [Google Scholar] [CrossRef]
- Hohenester, E. Laminin G-like domains: Dystroglycan-specific lectins. Curr. Opin. Struct. Biol. 2019, 56, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zwolanek, D.; Hu, Y.; Schreiber, G.; Brockmann, K.; Izu, Y.; Tian, Z.; Devoto, M.; Gandhy, S.; Meier, M.; et al. O.2 Collagen type XII: A new congenital matrix and muscle disease. Neuromuscul. Disord. 2013, 23, 739–740. [Google Scholar] [CrossRef]
- Neuhaus, S.; Konersman, C.; Saade, D.; Donkervoort, S.; Ceulemans, S.; Magoulas, P.; Skalsky, A.; Friedman, J.; Malicki, D.; Bainbridge, M.; et al. P.382Recessive COL12A1 loss of function EDS/myopathy overlap syndrome: Confirmation and expansion of a consistently severe phenotype. Neuromuscul. Disord. 2019, 29, S193. [Google Scholar] [CrossRef]
- Araújo, D.; Antunes, H. A novel mutation in the COL12A1 gene. Gene 2020, 768, 145266. [Google Scholar] [CrossRef]
- Cazzato, D.; Castori, M.; Lombardi, R.; Caravello, F.; Bella, E.D.; Petrucci, A.; Grammatico, P.; Dordoni, C.; Colombi, M.; Lauria, G. Small fiber neuropathy is a common feature of Ehlers-Danlos syndromes. Neurology 2016, 87, 155–159. [Google Scholar] [CrossRef]
- Voortman, M.; Fritz, D.; Vogels, O.J.M.; Eftimov, F.; Van De Beek, D.; Brouwer, M.C.; Drent, M. Small fiber neuropathy: A disabling and underrecognized syndrome. Curr. Opin. Pulm. Med. 2017, 23, 447–457. [Google Scholar] [CrossRef]
- Dusanic, M.; Dekomien, G.; Lücke, T.; Vorgerd, M.; Weis, J.; Epplen, J.T.; Köhler, C.; Hoffjan, S. Novel Nonsense Mutation in SLC39A13 Initially Presenting as Myopathy: Case Report and Review of the Literature. Mol. Syndromol. 2018, 9, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Izu, Y.; Sun, M.; Zwolanek, D.; Veit, G.; Williams, V.; Cha, B.; Jepsen, K.J.; Koch, M.; Birk, D.E. Type XII collagen regulates osteoblast polarity and communication during bone formation. J. Cell Biol. 2011, 193, 1115–1130. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Finding/Maneuver | Score | Patient |
|---|---|---|
| Hyperextension of the elbows beyond 10°. | ||
| Right | 1 | 1 |
| Left | 1 | 1 |
| Hyperextension of the knees beyond 10°. | ||
| Right | 1 | 1 |
| Left | 1 | 1 |
| Passive apposition of the thumbs to the flexor aspects of the forearms. | ||
| Right | 1 | 1 |
| Left | 1 | 1 |
| Passive dorsiflexion of the little fingers beyond 90°. | ||
| Right | 1 | 0 |
| Left | 1 | 0 |
| Forward flexion of the trunk, with knees fully extended, so that the palms of the hands rest easily on the floor. | 1 | 0 |
| Maximum score | 9 | 6 |
| Clinical Subtype | Abbreviation | Old Nomenclature | Inheritance Pattern | Gene | Protein | Predominant Clinical Features |
|---|---|---|---|---|---|---|
| Classical EDS | cEDS | Classical EDS, types I and II | AD | COL5A1, COL5A2, rarely COL1A1 | Type V collagen Type I collagen | Hyperextensible skin, atrophic scars, fragile skin, increased bruisability, doughy/velvety skin, and generalized joint hypermobility |
| Classical-like EDS | clEDS | TNXB-deficient EDS | AR | TNXB | Tenascin XB | Hyperextensible skin, velvety skin texture, no atrophic scarring, generalized joint hypermobility, and easy bruisability |
| Cardiac-valvular | cvEDS | AR | COL1A1 | Type I collagen | Progressive cardiac valve involvement, hyperextensible thin skin, atrophic scars, increased bruisability, and joint hypermobility | |
| Vascular EDS | vEDS | Vascular EDS, type IV | AD | COL3A1 | Type III collagen Type I collagen | Arterial rupture, internal organ rupture (colon, uterus), severe bruising, thin translucent skin, and small joint hypermobility |
| Hypermobile EDS | hEDS | Hypermobile EDS, type III | AD | Unknown | Unknown | Generalized joint hypermobility, mildly hyperextensible skin, soft velvety skin, recurrent hernias, organ prolapse, unexplained striae, chronic pain, and joint dislocations/subluxations |
| Arthrochalasia EDS | aEDS | Arthrochalasia, types VIIA and VIIB | AD | COL1A1, COL1A2 | Type I collagen | Congenital bilateral hip dislocation, severe generalized joint hypermobility, hyperextensible skin, tissue fragility, hypotonia, and mild osteopenia |
| Dermatosparaxis EDS | dEDS | Dermatosparaxis EDS, type VIIC | AR | ADAMTS2 | ADAMTS-2 | Severe skin fragility, visceral fragility, lax redundant skin, severe bruisability, and postnatal growth retardation |
| Kyphoscoliotic EDS | kEDS | Kyphoscoliosis EDS, type VI | AR | PLOD1 FKBP14 | LH1 FKBP22 | Congenital hypotonia, early kyphoscoliosis, generalized joint hypermobility, osteopenia, blue sclerae, and marfanoid habitus |
| Brittle Cornea Syndrome | BCS | Brittle cornea syndrome | AR | ZNF469 PRDM5 | ZNF469 PRDM5 | Thin cornea, keratoconus, blue sclerae, the risk for globe rupture, retinal detachment, and high myopia |
| Spondylodysplastic EDS | spEDS | EDS progeroid type Spondylocheirodysplastic EDS | AR | B4GALT7 B3GALT6 SLC39A13 | β4GalT7 β3GalT6 ZIP13 | Short stature, hypotonia, limb bowing, characteristic skeletal findings, osteopenia, hyperextensible, and thin doughy skin |
| Musculocontractural EDS | mcEDS | Adducted thumb Clubfoot Syndrome B3GalT6-deficient EDS EDS Kosho type | AR | CHST14 DSE | D4ST1 DSE | Congenital contractures (clubfoot), hyperextensible skin, easy bruisability, fragile skin, atrophic scars, and recurrent dislocations |
| Myopathic EDS | mEDS | AD or AR | COL12A1 | Type XII collagen | Congenital hypotonia, proximal joint contractures, distal joint hypermobility, doughy skin, and atrophic scars | |
| Periodontal EDS | pEDS | EDS periodontitis, type VIII | AD | C1R C1S | C1r C1s | Early-onset severe periodontitis, unattached gingiva, pretibial plaques, joint hypermobility, hyperextensible skin, and marfanoid features |
| The Five-Point Questionnaire |
|---|
| 1. Can you now (or could you ever) place your hands flat on the floor without bending your knees? |
| 2. Can you now (or could you ever) bend your thumb to touch your forearm? |
| 3. As a child, did you amuse your friends by contorting your body into strange shapes or could you do the splits? |
| 4. As a child or teenager, did your shoulder or kneecap dislocate on more than one occasion? |
| 5. Do you consider yourself “double-jointed”? |
| A “yes” answer to two or more questions suggests joint hypermobility with 80–85% sensitivity and 80–90% specificity |
| Genetic Variant Information | Allele Frequency | Pathogenicity Predictors | GT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chr | Ref | Alt | dbSNP | Change | 1000G | ExAC | gnomAD | SIFT | PolyPhen2 | PROVEAN | CADD | |
| COL12A1 | 6 | C | T | rs201988277 | exon51 c.C7853T p. T2618M | 0.0002 | 0.0003 | 0.0003 | D | D | D | 25.2 | 0/1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajardo-Jiménez, M.J.; Tejada-Moreno, J.A.; Mejía-García, A.; Villegas-Lanau, A.; Zapata-Builes, W.; Restrepo, J.E.; Cuartas, G.P.; Hernandez, J.C. Ehlers-Danlos: A Literature Review and Case Report in a Colombian Woman with Multiple Comorbidities. Genes 2022, 13, 2118. https://doi.org/10.3390/genes13112118
Fajardo-Jiménez MJ, Tejada-Moreno JA, Mejía-García A, Villegas-Lanau A, Zapata-Builes W, Restrepo JE, Cuartas GP, Hernandez JC. Ehlers-Danlos: A Literature Review and Case Report in a Colombian Woman with Multiple Comorbidities. Genes. 2022; 13(11):2118. https://doi.org/10.3390/genes13112118
Chicago/Turabian StyleFajardo-Jiménez, María José, Johanna A. Tejada-Moreno, Alejandro Mejía-García, Andrés Villegas-Lanau, Wildeman Zapata-Builes, Jorge E. Restrepo, Gina P. Cuartas, and Juan C. Hernandez. 2022. "Ehlers-Danlos: A Literature Review and Case Report in a Colombian Woman with Multiple Comorbidities" Genes 13, no. 11: 2118. https://doi.org/10.3390/genes13112118
APA StyleFajardo-Jiménez, M. J., Tejada-Moreno, J. A., Mejía-García, A., Villegas-Lanau, A., Zapata-Builes, W., Restrepo, J. E., Cuartas, G. P., & Hernandez, J. C. (2022). Ehlers-Danlos: A Literature Review and Case Report in a Colombian Woman with Multiple Comorbidities. Genes, 13(11), 2118. https://doi.org/10.3390/genes13112118