

Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias

Abstract
:1. Introduction
2. Subjects and Methods
2.1. Patients and Study Design
2.2. DNA Analysis
2.3. In Silico Protein Structure Analysis
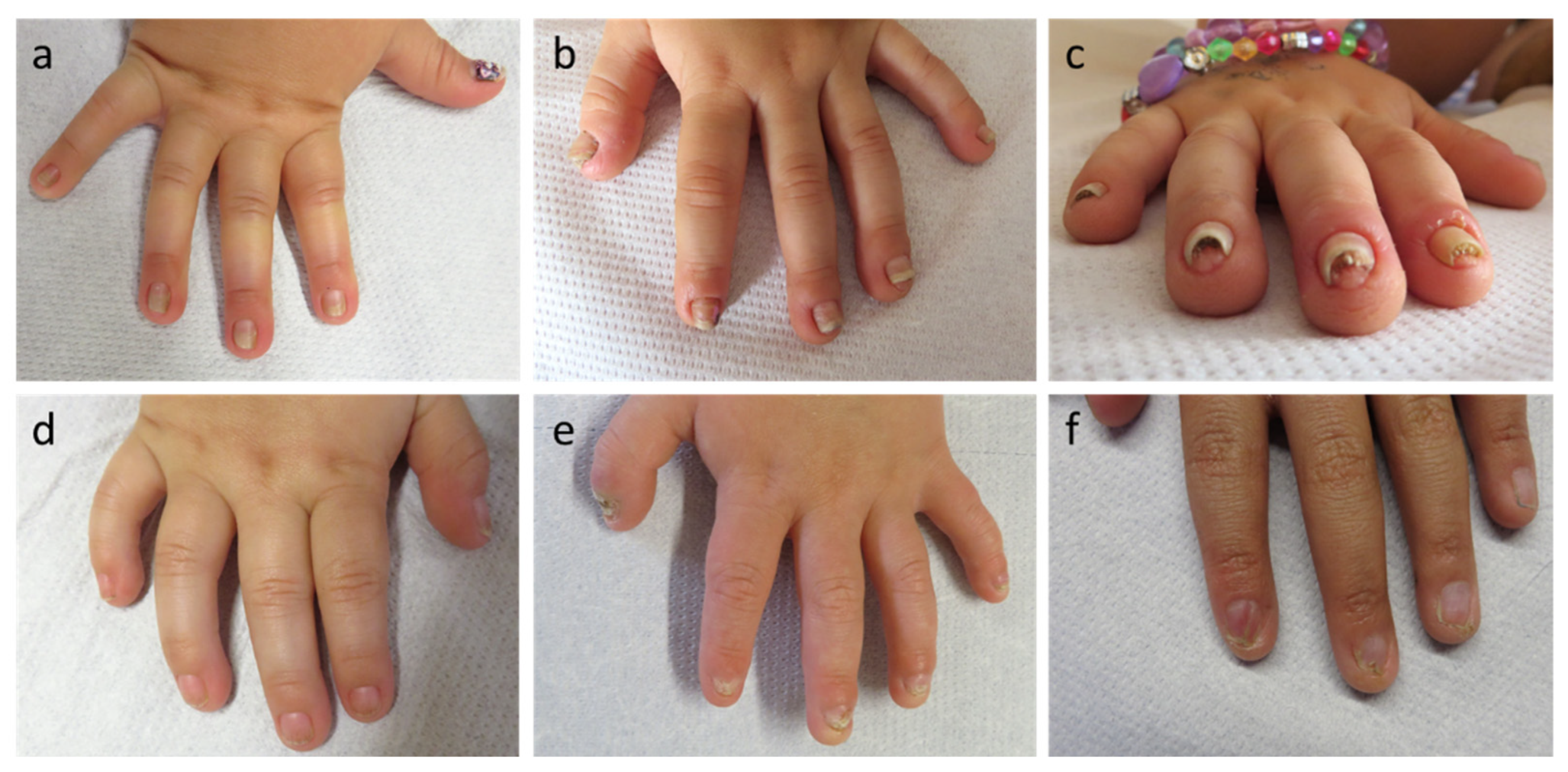
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piraccini, B.M.; Starace, M. Nail disorders in infants and children. Curr. Opin. Pediatr. 2014, 26, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Starace, M.; Alessandrini, A.; Piraccini, B.M. Nail disorders in children. Skin Appendage Disord. 2018, 4, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, A.; Tamayo, L.; Sosa-de-Martínez, C.; Durán-McKinster, C.; Orozco-Covarrubias, L.; Ruiz-Maldonado, R. Prevalence and nature of nail alterations in pediatric patients. Pediatr. Dermatol. 2001, 18, 107–109. [Google Scholar] [CrossRef]
- Tasia, M.; Lecerf, P.; Richert, B.; André, J. Paediatric nail consultation in an academic centre in Belgium: A 10-year retrospective study. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1800–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzberg, A.J. Nail manifestations of systemic diseases. Clin. Podiatr. Med. Surg. 1995, 12, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar] [CrossRef]
- Monreal, A.W.; Ferguson, B.M.; Headon, D.J.; Street, S.L.; Overbeek, P.A.; Zonana, J. Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat. Genet. 1999, 22, 366–369. [Google Scholar] [CrossRef]
- Kohn, L.L.; Braun, M.; Cordoro, K.M.; McCalmont, T.H.; Shah, S.D.; Frieden, I.J.; Mathur, A.N. Skin and mucosal manifestations in NEMO syndrome: A case series and literature review. Pediatr. Dermatol. 2022, 39, 84–90. [Google Scholar] [CrossRef]
- Bohring, A.; Stamm, T.; Spaich, C.; Haase, C.; Spree, K.; Hehr, U.; Hoffmann, M.; Ledig, S.; Sel, S.; Wieacker, P.; et al. WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am. J. Hum. Genet. 2009, 85, 97–105. [Google Scholar] [CrossRef]
- Krøigård, A.B.; Clemmensen, O.; Gjørup, H.; Hertz, J.M.; Bygum, A. Odonto-onycho-dermal dysplasia in a patient homozygous for a WNT10A nonsense mutation and mild manifestations of ectodermal dysplasia in carriers of the mutation. BMC Dermatol. 2016, 16, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.C.; Lee, J.Y.; Hsu, M.M.; Chao, S.C. Case report of Schöpf-Schulz-Passarge syndrome resulting from a missense mutation, p.Arg104Cys, in WNT10A. J. Dermatol. 2018, 45, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Kibar, Z.; Der Kaloustian, V.M.; Brais, B.; Hani, V.; Fraser, F.C.; Rouleau, G.A. The gene responsible for Clouston hidrotic ectodermal dysplasia maps to the pericentromeric region of chromosome 13q. Hum. Mol. Genet. 1996, 5, 543–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, S.M.; de Jong, T.P.; Buss, P.; Hennekam, R.C. EEC syndrome and genitourinary anomalies: An update. Am. J. Med. Genet. 1996, 63, 472–478. [Google Scholar] [CrossRef]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Ferstl, P.; Wohlfart, S.; Schneider, H. Sweating ability of patients with p63-associated syndromes. Eur. J. Pediatr. 2018, 177, 1727–1731. [Google Scholar] [CrossRef]
- Littman, A.; Levin, S. Anonychia as a recessive autosomal trait in man. J. Investig. Dermatol. 1964, 42, 177–178. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; Senderek, J.; Anhuf, D.; Thiel, C.T.; Ekici, A.B.; Poblete-Gutierrez, P.; van Steensel, M.; Seelow, D.; Nürnberg, G.; Schild, H.H.; et al. Mutations in the gene encoding the Wnt-signaling component R-spondin 4 (RSPO4) cause autosomal recessive anonychia. Am. J. Hum. Genet. 2006, 79, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Brüchle, N.O.; Frank, J.; Frank, V.; Senderek, J.; Akar, A.; Koc, E.; Rigopoulos, D.; van Steensel, M.; Zerres, K.; Bergmann, C. RSPO4 is the major gene in autosomal-recessive anonychia and mutations cluster in the furin-like cysteine-rich domains of the Wnt signaling ligand R-spondin 4. J. Investig. Dermatol. 2008, 128, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Blaydon, D.C.; Ishii, Y.; O’Toole, E.A.; Unsworth, H.C.; Teh, M.T.; Rüschendorf, F.; Sinclair, C.; Hopsu-Havu, V.K.; Tidman, N.; Moss, C.; et al. The gene encoding R-spondin 4 (RSPO4), a secreted protein implicated in Wnt signaling, is mutated in inherited anonychia. Nat. Genet. 2006, 38, 1245–1247. [Google Scholar] [CrossRef]
- Hsu, C.K.; Romano, M.T.; Nanda, A.; Rashidghamat, E.; Lee, J.Y.W.; Huang, H.Y.; Songsantiphap, C.; Lee, J.Y.; Al-Ajmi, H.; Betz, R.C.; et al. Congenital anonychia and uncombable hair syndrome: Coinheritance of homozygous mutations in RSPO4 and PADI3. J. Investig. Dermatol. 2017, 137, 1176–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlfart, S.; Hammersen, J.; Schneider, H. Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements. J. Hum. Genet. 2016, 61, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Raza, S.I.; Navid, A.K.; Noor, Z.; Shah, K.; Dar, N.R.; Ahmad, W.; Rashid, S. Gly67Arg substitution in RSPO4 disrupts the WNT signaling pathway due to an abnormal binding pattern with LGRs leading to anonychia. RSC Adv. 2017, 7, 17357–17366. [Google Scholar] [CrossRef] [Green Version]
- Bloom, A.; Blanken, B.; Schlakman, B.; Arena, T.; Mironov, Z.; Vlahovic, T.C. A review of nail dystrophies for the practitioner. Adv. Skin Wound Care 2020, 33, 20–26. [Google Scholar] [CrossRef]
- Belyayeva, E.; Gregoriou, S.; Chalikias, J.; Kontochristopoulos, G.; Koumantaki, E.; Makris, M.; Koti, I.; Katoulis, A.; Katsambas, A.; Rigopoulos, D. The impact of nail disorders on quality of life. Eur. J. Dermatol. 2013, 23, 366–371. [Google Scholar] [CrossRef]
- Nakata, M.; Koshiba, H.; Eto, K.; Nance, W.E. A genetic study of anodontia in X-linked hypohidrotic ectodermal dysplasia. Am. J. Hum. Genet. 1980, 32, 908–919. [Google Scholar]
- Mortier, K.; Wackens, G. Ectodermal dysplasia anhidrotic. Orphanet Encycl. 2004, 3, 1–6. [Google Scholar]
- Fete, M.; Hermann, J.; Behrens, J.; Huttner, K.M. X-linked hypohidrotic ectodermal dysplasia (XLHED): Clinical and diagnostic insights from an international patient registry. Am. J. Med. Genet. Part A 2014, 164, 2437–2442. [Google Scholar] [CrossRef]
- Bellet, J.S. Pediatric nail disorders. Dermatol. Clin. 2021, 39, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Betz, R.C. Nails—More than just an ectodermal appendage: The genetics behind isolated nail disorders. Br. J. Dermatol. 2015, 173, 886. [Google Scholar] [CrossRef] [PubMed]
- Fröjmark, A.S.; Schuster, J.; Sobol, M.; Entesarian, M.; Kilander, M.B.C.; Gabrikova, D.; Nawaz, S.; Baig, S.M.; Schulte, G.; Klar, J.; et al. Mutations in Frizzled 6 cause isolated autosomal-recessive nail dysplasia. Am. J. Hum. Genet. 2011, 88, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Basit, S.; Habib, R.; Kamal, A.; Muhammad, N.; Ahmad, W. Genetics of human isolated hereditary nail disorders. Br. J. Dermatol. 2015, 173, 922–929. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease(s) | Affected Gene(s) | Number of Patients Seen 2017–2022 | Patients with Nail Abnormalities |
|---|---|---|---|
| Hypohidrotic ED, X-linked | EDA | 161 | 0 (0%) |
| Hypohidrotic ED, autosomal dominant or recessive | EDAR, EDARADD | 14 | 2 (14.3%) |
| Incontinentia pigmenti | IKBKG | 3 | 0 (0%) |
| Odonto-onycho-dermal dysplasia/ Schöpf-Schulz-Passarge syndrome | WNT10A | 16 | 15 (93.8%) |
| Clouston syndrome | GJB6 2 | 2 | 2 (100%) |
| Ankyloblepharon-ED-cleft lip/palate (AEC) syndrome | TP63 | 4 | 4 (100%) |
| Ectrodactyly-ED-cleft lip/palate (EEC) syndrome | TP63 | 3 | 2 (66.7%) |
| Anonychia congenita | RSPO4 | 1 | 1 (100%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier-Wohlfart, S.; Aicher, C.; Willershausen, I.; Peschel, N.; Meißner, U.; Gölz, L.; Schneider, H. Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias. Genes 2022, 13, 2119. https://doi.org/10.3390/genes13112119
Maier-Wohlfart S, Aicher C, Willershausen I, Peschel N, Meißner U, Gölz L, Schneider H. Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias. Genes. 2022; 13(11):2119. https://doi.org/10.3390/genes13112119
Chicago/Turabian StyleMaier-Wohlfart, Sigrun, Carmen Aicher, Ines Willershausen, Nicolai Peschel, Udo Meißner, Lina Gölz, and Holm Schneider. 2022. "Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias" Genes 13, no. 11: 2119. https://doi.org/10.3390/genes13112119
APA StyleMaier-Wohlfart, S., Aicher, C., Willershausen, I., Peschel, N., Meißner, U., Gölz, L., & Schneider, H. (2022). Congenital Nail Disorders among Children with Suspected Ectodermal Dysplasias. Genes, 13(11), 2119. https://doi.org/10.3390/genes13112119