
Challenges with Simulating Modified RNA: Insights into Role and Reciprocity of Experimental and Computational Approaches


Abstract
:1. Introduction
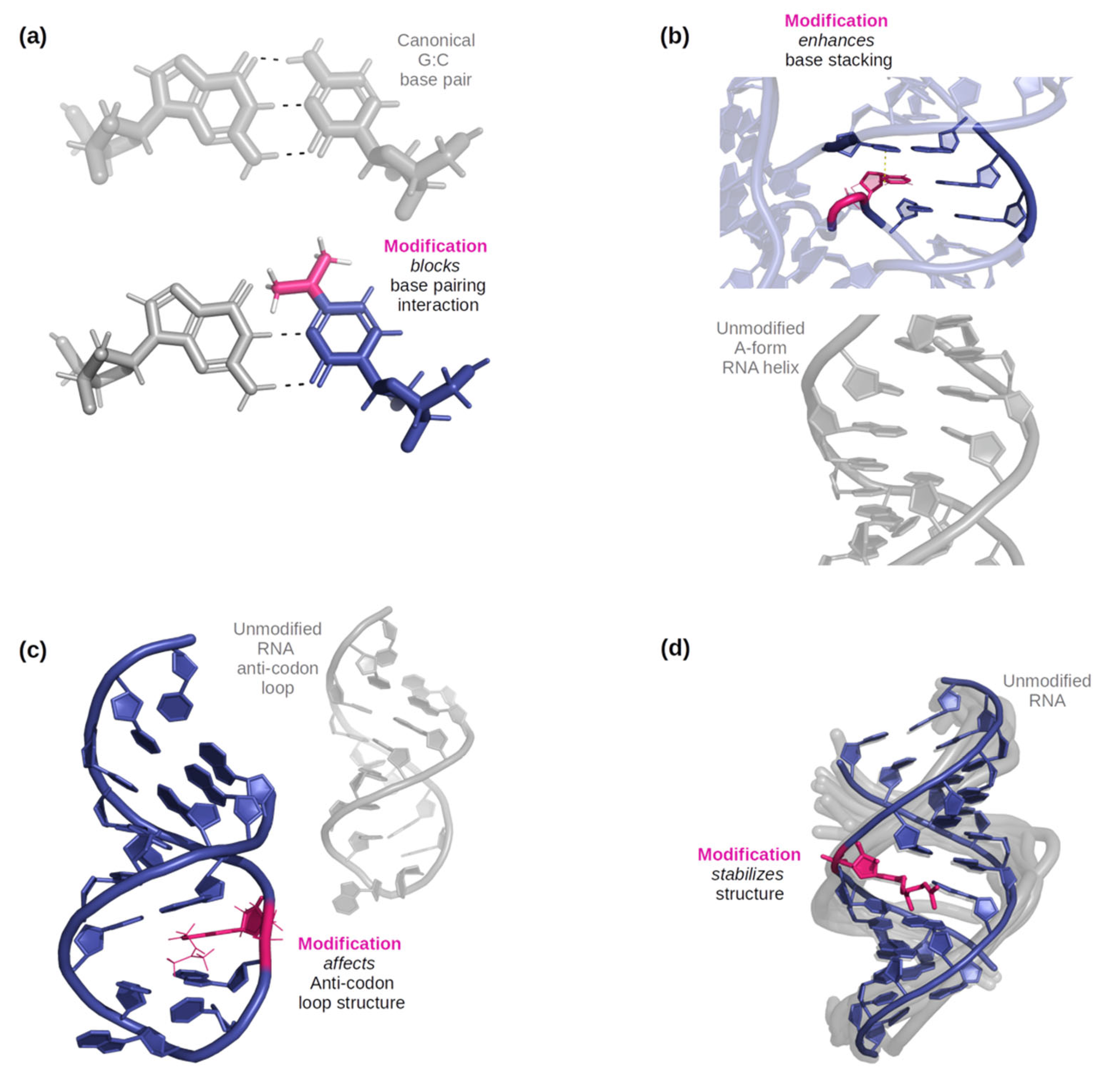
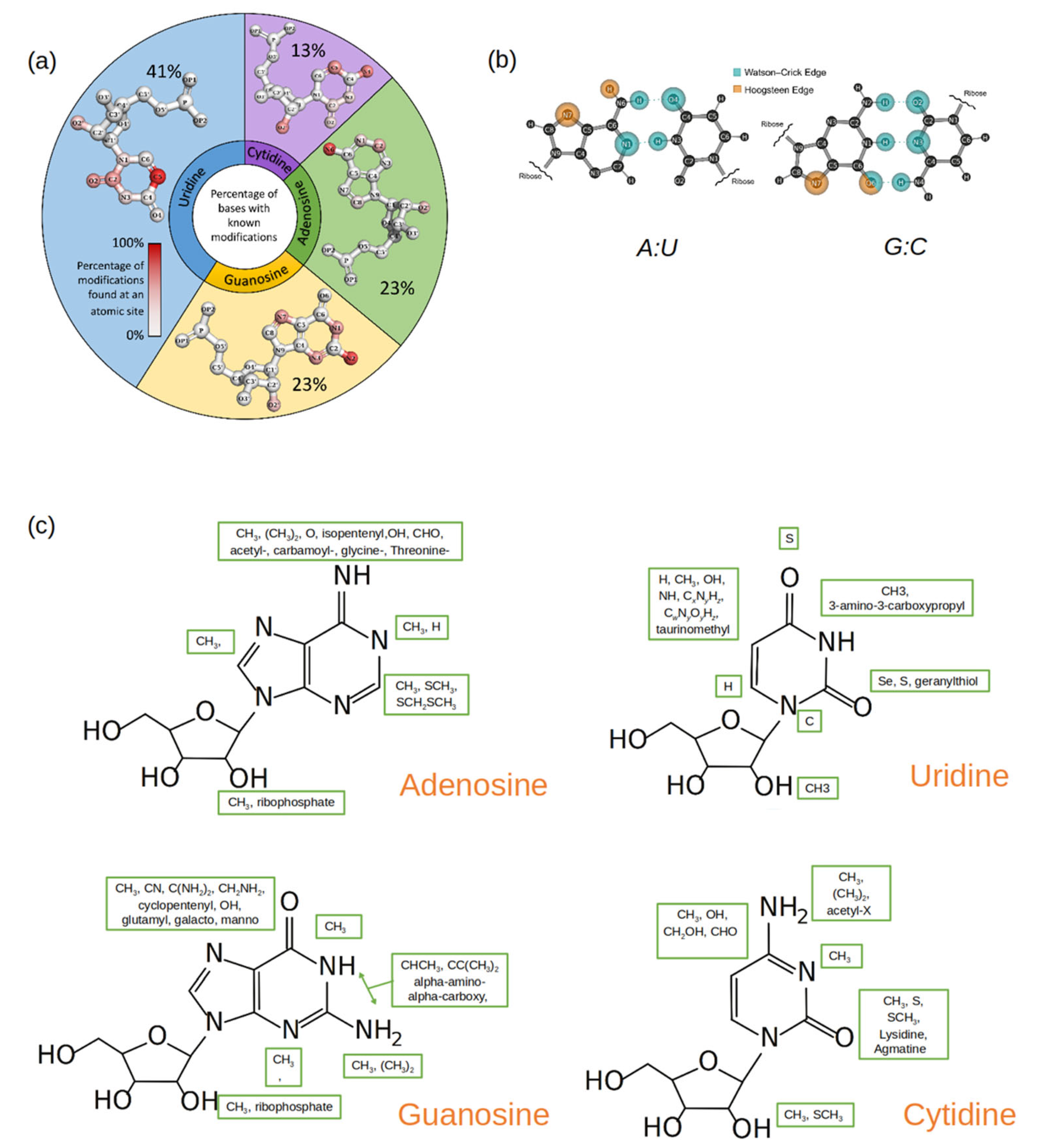
2. Classification of Modified RNA Nucleosides Based on Their Structural/Functional Implications
2.1. Based on the Location of the Modified Group in the Modified Nucleotide
2.2. Based on the Nature of the Modified Group in the Modified Nucleotide
2.3. Summary of the Classification of RNA Modifications
3. Molecular Dynamics Simulations (MDS) of Modified RNA
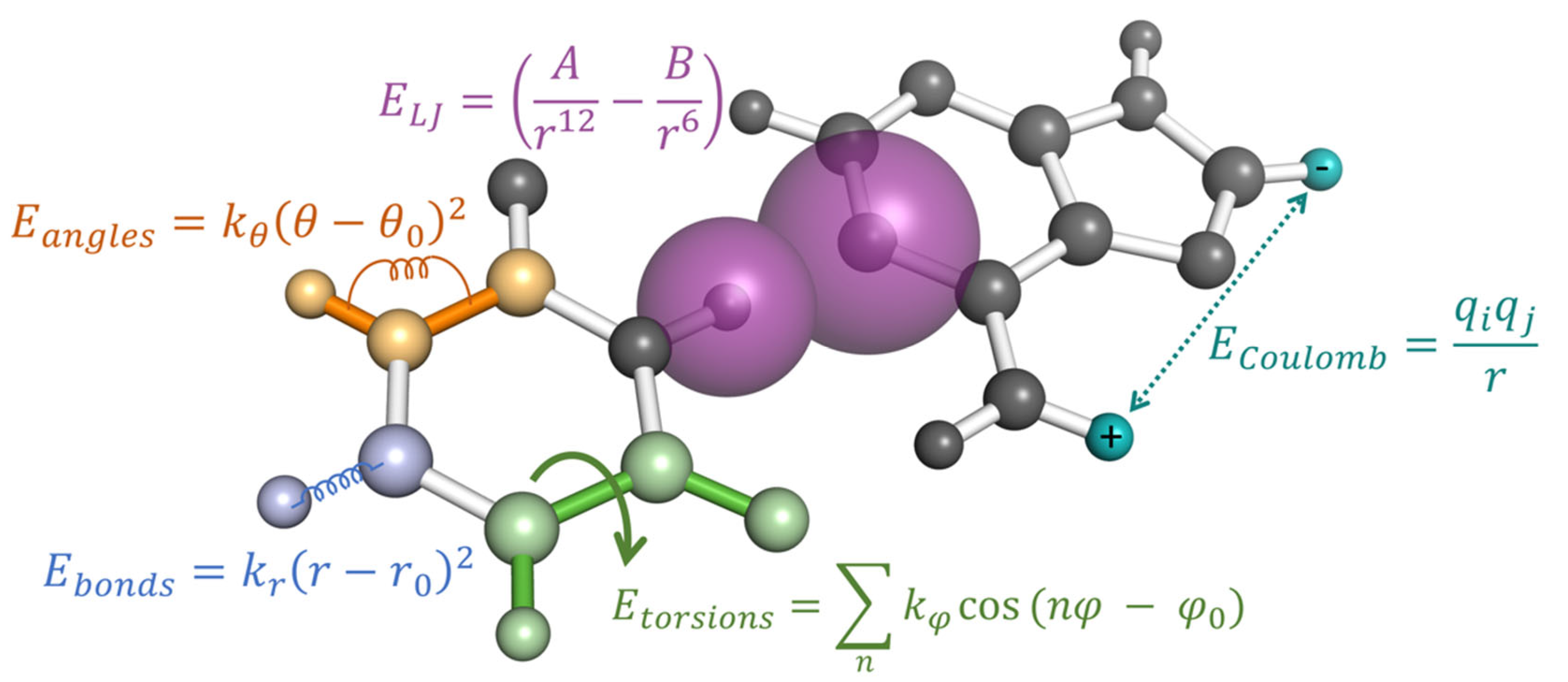
3.1. Force-Field Parameterization Strategies
3.2. Modified Nucleotide Parameterization Strategies for AMBER and CHARMM
3.3. Molecular Dynamics Simulation Studies of Modified RNA
3.4. Summary of MDS of Modified RNA
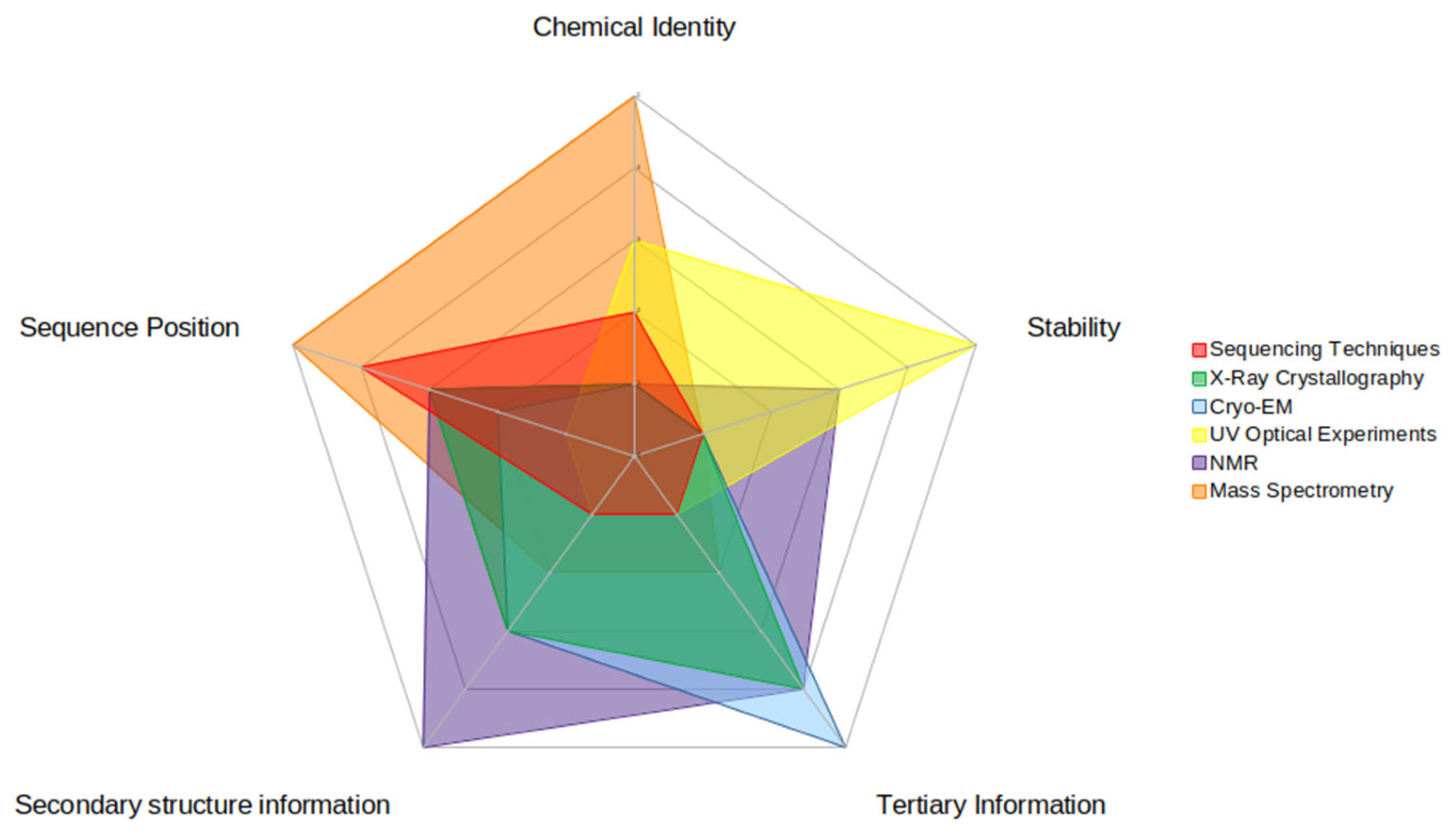
4. Experiments That Can Help Validate MD Simulation Results
4.1. Detection Methods
4.1.1. Mass Spectrometry (MS)
4.1.2. Sequencing Techniques
4.2. Structural Analysis Methods
4.2.1. UV Optical Melting Experiments
4.2.2. Nuclear Magnetic Resonance (NMR)
4.2.3. X-ray Crystallography
4.2.4. Cryogenic Electron Microscopy (Cryo-EM)
4.3. Summary of Experimental Approaches for Modified RNA Research
5. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCown, P.J.; Ruszkowska, A.; Kunkler, C.N.; Breger, K.; Hulewicz, J.P.; Wang, M.C.; Springer, N.A.; Brown, J.A. Naturally Occurring Modified Ribonucleosides. Wiley Interdiscip. Rev. RNA 2020, 11, e1595. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. TRNA Stabilization by Modified Nucleotides. Biochemistry 2010, 49, 4934–4944. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The Emerging Role of RNA Modifications in the Regulation of MRNA Stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA Modifications: Form, Distribution, and Function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrino, S.; Dent, K.C.; Spikes, T.; Warren, A.J. Cryo-EM Reconstruction of the Human 40S Ribosomal Subunit at 2.15 Å Resolution. bioRxiv 2022. [Google Scholar] [CrossRef]
- Maden, B.E. The Numerous Modified Nucleotides in Eukaryotic Ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol. 1990, 39, 241–303. [Google Scholar]
- Höfer, K.; Jäschke, A. Epitranscriptomics: RNA Modifications in Bacteria and Archaea. Microbiol Spectr 2018, 6, 3. [Google Scholar] [CrossRef]
- Marbaniang, C.N.; Vogel, J. Emerging Roles of RNA Modifications in Bacteria. Curr. Opin. Microbiol. 2016, 30, 50–57. [Google Scholar] [CrossRef]
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol. Med. 2018, 24, 886–903. [Google Scholar] [CrossRef]
- Gokhale, N.S.; Horner, S.M. RNA Modifications Go Viral. PLoS Pathog. 2017, 13, e1006188. [Google Scholar] [CrossRef]
- Eyer, L.; Nencka, R.; de Clercq, E.; Seley-Radtke, K.; Růžek, D. Nucleoside Analogs as a Rich Source of Antiviral Agents Active against Arthropod-Borne Flaviviruses. Antivir. Chem. Chemother. 2018, 26, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The Current Landscape of Nucleic Acid Therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Weissman, D. Nucleoside Modified MRNA Vaccines for Infectious Diseases. Methods Mol. Biol. 2017, 1499, 109–121. [Google Scholar]
- Cohn, W.E.; Volkin, E. Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature 1951, 167, 483–484. [Google Scholar] [CrossRef]
- Schaefer, M.; Kapoor, U.; Jantsch, M.F. Understanding RNA Modifications: The Promises and Technological Bottlenecks of the ‘Epitranscriptome’. Open Biol. 2017, 7, 170077. [Google Scholar] [CrossRef] [Green Version]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-Wide Mapping of 5-Methylcytidine RNA Modifications in Bacteria, Archaea, and Yeast Reveals M5C within Archaeal MRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-Wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of NcRNA and MRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiong, X.; Wang, K.; Wang, L.; Shu, X.; Ma, S.; Yi, C. Transcriptome-Wide Mapping Reveals Reversible and Dynamic N(1)-Methyladenosine Methylome. Nat. Chem. Biol. 2016, 12, 311–316. [Google Scholar] [CrossRef]
- Wilkinson, E.; Cui, Y.-H.; He, Y.-Y. Context-Dependent Roles of RNA Modifications in Stress Responses and Diseases. Int. J. Mol. Sci. 2021, 22, 1949. [Google Scholar] [CrossRef]
- Dedon, P.C.; Begley, T.J. A System of RNA Modifications and Biased Codon Use Controls Cellular Stress Response at the Level of Translation. Chem. Res. Toxicol. 2014, 27, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Satterlee, J.S.; Basanta-Sanchez, M.; Blanco, S.; Li, J.B.; Meyer, K.; Pollock, J.; Sadri-Vakili, G.; Rybak-Wolf, A. Novel RNA Modifications in the Nervous System: Form and Function. J. Neurosci. 2014, 34, 15170–15177. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, K.; Ding, G.; Huang, H. Advances in Research into Gamete and Embryo-Fetal Origins of Adult Diseases. Sci. China Life Sci. 2019, 62, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Westhof, E.; Fritsch, V. RNA Folding: Beyond Watson-Crick Pairs. Structure 2000, 8, R55–R65. [Google Scholar] [CrossRef] [Green Version]
- Batey, R.T.; Rambo, R.P.; Doudna, J.A. Tertiary Motifs in RNA Structure and Folding. Angew. Chem. Int. Ed Engl. 1999, 38, 2326–2343. [Google Scholar] [CrossRef]
- Lemieux, S.; Major, F. RNA Canonical and Non-Canonical Base Pairing Types: A Recognition Method and Complete Repertoire. Nucleic Acids Res. 2002, 30, 4250–4263. [Google Scholar] [CrossRef] [Green Version]
- Jhunjhunwala, A.; Ali, Z.; Bhattacharya, S.; Halder, A.; Mitra, A.; Sharma, P. On the Nature of Nucleobase Stacking in RNA: A Comprehensive Survey of Its Structural Variability and a Systematic Classification of Associated Interactions. J. Chem. Inf. Model. 2021, 61, 1470–1480. [Google Scholar] [CrossRef]
- Baulin, E.; Metelev, V.; Bogdanov, A. Base-Intercalated and Base-Wedged Stacking Elements in 3D-Structure of RNA and RNA-Protein Complexes. Nucleic Acids Res. 2020, 48, 8675–8685. [Google Scholar] [CrossRef]
- Chawla, M.; Oliva, R.; Bujnicki, J.M.; Cavallo, L. An Atlas of RNA Base Pairs Involving Modified Nucleobases with Optimal Geometries and Accurate Energies. Nucleic Acids Res. 2015, 43, 6714–6729. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Stefaniak, F.; Ray, A.; Cappannini, A.; Mukherjee, S.; Purta, E.; Kurkowska, M.; Shirvanizadeh, N.; Destefanis, E.; Groza, P.; et al. MODOMICS: A Database of RNA Modification Pathways. 2021 Update. Nucleic Acids Res. 2022, 50, D231–D235. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-Methyladenosine-Dependent RNA Structural Switches Regulate RNA-Protein Interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Bohleber, S.; Fradejas-Villar, N. The Modified Base Isopentenyladenosine and Its Derivatives in TRNA. RNA Biol. 2017, 14, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirci, H.; Murphy, F., 4th; Belardinelli, R.; Kelley, A.C.; Ramakrishnan, V.; Gregory, S.T.; Dahlberg, A.E.; Jogl, G. Modification of 16S Ribosomal RNA by the KsgA Methyltransferase Restructures the 30S Subunit to Optimize Ribosome Function. RNA 2010, 16, 2319–2324. [Google Scholar] [CrossRef] [Green Version]
- Rife, J.P.; Cheng, C.S.; Moore, P.B.; Strobel, S.A. N 2-Methylguanosine Is Iso-Energetic with Guanosine in RNA Duplexes and GNRA Tetraloops. Nucleic Acids Res. 1998, 26, 3640–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallan, P.S.; Kreutz, C.; Bosio, S.; Micura, R.; Egli, M. Effects of N2,N2-Dimethylguanosine on RNA Structure and Stability: Crystal Structure of an RNA Duplex with Tandem M2 2G:A Pairs. RNA 2008, 14, 2125–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, S.; Sekula, B.; Ruszkowski, M.; Ranganathan, S.V.; Haruehanroengra, P.; Wu, Y.; Shen, F.; Sheng, J. Base Pairing, Structural and Functional Insights into N4-Methylcytidine (M4C) and N4,N4-Dimethylcytidine (M42C) Modified RNA. Nucleic Acids Res. 2020, 48, 10087–10100. [Google Scholar] [CrossRef]
- Shi, H.; Moore, P.B. The Crystal Structure of Yeast Phenylalanine TRNA at 1.93 A Resolution: A Classic Structure Revisited. RNA 2000, 6, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Denmon, A.P.; Wang, J.; Nikonowicz, E.P. Conformation Effects of Base Modification on the Anticodon Stem-Loop of Bacillus Subtilis TRNA(Tyr). J. Mol. Biol. 2011, 412, 285–303. [Google Scholar] [CrossRef]
- Wang, R.; Vangaveti, S.; Ranganathan, S.V.; Basanta-Sanchez, M.; Haruehanroengra, P.; Chen, A.; Sheng, J. Synthesis, Base Pairing and Structure Studies of Geranylated RNA. Nucleic Acids Res. 2016, 44, 6036–6045. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Luo, Z.; He, K.; Delaney, M.O.; Chen, D.; Sheng, J. Base Pairing and Structural Insights into the 5-Formylcytosine in RNA Duplex. Nucleic Acids Res. 2016, 44, 4968–4977. [Google Scholar] [CrossRef] [Green Version]
- Auxilien, S.; Rasmussen, A.; Rose, S.; Brochier-Armanet, C.; Husson, C.; Fourmy, D.; Grosjean, H.; Douthwaite, S. Specificity Shifts in the RRNA and TRNA Nucleotide Targets of Archaeal and Bacterial M5U Methyltransferases. RNA 2011, 17, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, Y.; Kimura, S.; Suzuki, T. Dual Pathways of TRNA Hydroxylation Ensure Efficient Translation by Expanding Decoding Capability. Nat. Commun. 2019, 10, 2858. [Google Scholar] [CrossRef]
- Weissenbach, J.; Dirheimer, G. Pairing Properties of the Methylester of 5-Carboxymethyl Uridine in the Wobble Position of Yeast TRNA3Arg. Biochim. Biophys. Acta 1978, 518, 530–534. [Google Scholar] [CrossRef]
- Nilsson, K.; Jäger, G.; Björk, G.R. An Unmodified Wobble Uridine in TRNAs Specific for Glutamine, Lysine, and Glutamic Acid from Salmonella Enterica Serovar Typhimurium Results in Nonviability-Due to Increased Missense Errors? PLoS ONE 2017, 12, e0175092. [Google Scholar]
- Dumelin, C.E.; Chen, Y.; Leconte, A.M.; Chen, Y.G.; Liu, D.R. Discovery and Biological Characterization of Geranylated RNA in Bacteria. Nat. Chem. Biol. 2012, 8, 913–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agris, P.F.; Eruysal, E.R.; Narendran, A.; Väre, V.Y.P.; Vangaveti, S.; Ranganathan, S.V. Celebrating Wobble Decoding: Half a Century and Still Much Is New. RNA Biol. 2018, 15, 537–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.C.; Hamlow, L.A.; Devereaux, Z.J.; Zhu, Y.; Nei, Y.-W.; Fan, L.; McNary, C.P.; Maitre, P.; Steinmetz, V.; Schindler, B.; et al. Structural and Energetic Effects of O2’-Ribose Methylation of Protonated Purine Nucleosides. J. Phys. Chem. B 2018, 122, 9147–9160. [Google Scholar] [CrossRef]
- Ontiveros, R.J.; Stoute, J.; Liu, K.F. The Chemical Diversity of RNA Modifications. Biochem. J 2019, 476, 1227–1245. [Google Scholar] [CrossRef]
- Elliott, B.A.; Ho, H.-T.; Ranganathan, S.V.; Vangaveti, S.; Ilkayeva, O.; Abou Assi, H.; Choi, A.K.; Agris, P.F.; Holley, C.L. Modification of Messenger RNA by 2′-O-Methylation Regulates Gene Expression in Vivo. Nat. Commun. 2019, 10, 3401. [Google Scholar] [CrossRef] [Green Version]
- Aström, S.U.; Byström, A.S. Rit1, a TRNA Backbone-Modifying Enzyme That Mediates Initiator and Elongator TRNA Discrimination. Cell 1994, 79, 535–546. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Wu, Y.; Begley, T.J.; Sheng, J. Sulfur Modification in Natural RNA and Therapeutic Oligonucleotides. RSC Chem. Biol. 2021, 2, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; He, C. Pseudouridine in a New Era of RNA Modifications. Cell Res. 2015, 25, 153–154. [Google Scholar] [CrossRef] [Green Version]
- Rozov, A.; Demeshkina, N.; Khusainov, I.; Westhof, E.; Yusupov, M.; Yusupova, G. Novel Base-Pairing Interactions at the TRNA Wobble Position Crucial for Accurate Reading of the Genetic Code. Nat. Commun. 2016, 7, 10457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, I.S.; Ng, C.L.; Kelley, A.C.; Wu, G.; Yu, Y.-T.; Ramakrishnan, V. Unusual Base Pairing during the Decoding of a Stop Codon by the Ribosome. Nature 2013, 500, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, B.A.; Kwon, S.Y.; Chamorro, M.; Oroszlan, S.; Hatfield, D.L.; Lee, B.J. Transfer RNA Modification Status Influences Retroviral Ribosomal Frameshifting. Virology 1999, 255, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumbhar, N.M.; Kumbhar, B.V.; Sonawane, K.D. Structural Significance of Hypermodified Nucleic Acid Base Hydroxywybutine (OHyW) Which Occur at 37th Position in the Anticodon Loop of Yeast TRNAPhe. J. Mol. Graph. Model. 2012, 38, 174–185. [Google Scholar] [CrossRef]
- Fandilolu, P.M.; Kamble, A.S.; Dound, A.S.; Sonawane, K.D. Role of Wybutosine and Mg2+ Ions in Modulating the Structure and Function of TRNAPhe: A Molecular Dynamics Study. ACS Omega 2019, 4, 21327–21339. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.A.; Pedram, K.; Malaker, S.A.; Batista, P.J.; Smith, B.A.H.; Johnson, A.G.; George, B.M.; Majzoub, K.; Villalta, P.W.; Carette, J.E.; et al. Small RNAs Are Modified with N-Glycans and Displayed on the Surface of Living Cells. Cell 2021, 184, 3109–3124.e22. [Google Scholar] [CrossRef]
- Nainytė, M.; Müller, F.; Ganazzoli, G.; Chan, C.-Y.; Crisp, A.; Globisch, D.; Carell, T. Amino Acid Modified RNA Bases as Building Blocks of an Early Earth RNA-Peptide World. Chemistry 2020, 26, 14856–14860. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Steinbrecher, T.; Latzer, J.; Case, D.A. Revised AMBER Parameters for Bioorganic Phosphates. J. Chem. Theory Comput. 2012, 8, 4405–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.A.; García, A.E. High-Resolution Reversible Folding of Hyperstable RNA Tetraloops Using Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. USA 2013, 110, 16820–16825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkul, J.A.; MacKerell, A.D., Jr. Polarizable Force Field for RNA Based on the Classical Drude Oscillator. J. Comput. Chem. 2018, 39, 2624–2646. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018, 118, 4177–4338. [Google Scholar] [CrossRef] [Green Version]
- Vangaveti, S.; Ranganathan, S.V.; Chen, A.A. Advances in RNA Molecular Dynamics: A Simulator’s Guide to RNA Force Fields. Wiley Interdiscip. Rev. RNA 2017, 8, e1396. [Google Scholar] [CrossRef]
- Xu, Y.; Vanommeslaeghe, K.; Aleksandrov, A.; MacKerell, A.D., Jr.; Nilsson, L. Additive CHARMM Force Field for Naturally Occurring Modified Ribonucleotides. J. Comput. Chem. 2016, 37, 896–912. [Google Scholar] [CrossRef] [Green Version]
- Aduri, R.; Psciuk, B.T.; Saro, P.; Taniga, H.; Schlegel, H.B.; SantaLucia, J. AMBER Force Field Parameters for the Naturally Occurring Modified Nucleosides in RNA. J. Chem. Theory Comput. 2007, 3, 1464–1475. [Google Scholar] [CrossRef]
- AMBER Parameter Database. Available online: http://amber.manchester.ac.uk/ (accessed on 27 January 2022).
- Prabhakar, P.S.; Takyi, N.A.; Wetmore, S.D. Posttranscriptional Modifications at the 37th Position in the Anticodon Stem–Loop of TRNA: Structural Insights from MD Simulations. RNA 2021, 27, 202–220. [Google Scholar] [CrossRef]
- Ten, G.N.; Yakovleva, A.A.; Nechaev, V.V.; Baranov, V.I. Hydrogen Bond Effect on the Structure and Vibrational Spectra of Complementary Pairs of Nucleic Acid Bases. III. Guanine-Cytosine. J. Struct. Chem. 2012, 53, 835–844. [Google Scholar] [CrossRef]
- Colarusso, P.; Zhang, K.; Guo, B.; Bernath, P.F. The Infrared Spectra of Uracil, Thymine, and Adenine in the Gas Phase. Chem. Phys. Lett. 1997, 269, 39–48. [Google Scholar] [CrossRef]
- Szczesniak, M.; Nowak, M.J.; Rostkowska, H.; Szczepaniak, K.; Person, W.B.; Shugar, D. Matrix Isolation Studies of Nucleic Acid Constituents. 1. Infrared Spectra of Uracil Monomers. J. Am. Chem. Soc. 1983, 105, 5969–5976. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M. Taking into Account the Ion-Induced Dipole Interaction in the Nonbonded Model of Ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kührová, P.; Otyepka, M.; Šponer, J.; Banáš, P. Are Waters around RNA More than Just a Solvent? - An Insight from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Oweida, T.J.; Kim, H.S.; Donald, J.M.; Singh, A.; Yingling, Y.G. Assessment of AMBER Force Fields for Simulations of SsDNA. J. Chem. Theory Comput. 2021, 17, 1208–1217. [Google Scholar] [CrossRef]
- Cassone, G.; Kruse, H.; Sponer, J. Interactions between Cyclic Nucleotides and Common Cations: An Ab initio Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2019, 21, 8121–8132. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Foloppe, N.; MacKerell, A.D., Jr. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Yin, D.; MacKerell, A.D. Combinedab initio/Empirical Approach for Optimization of Lennard-Jones Parameters. J. Comput. Chem. 1998, 19, 334–348. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Karplus, M. Importance of Attractive van Der Waals Contribution in Empirical Energy Function Models for the Heat of Vaporization of Polar Liquids. J. Phys. Chem. 1991, 95, 10559–10560. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. CHARMM Additive and Polarizable Force Fields for Biophysics and Computer-Aided Drug Design. Biochim. Biophys. Acta 2015, 1850, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKerell Lab. Available online: https://www.charmm.org/charmm/resources/charmm-force-fields/ (accessed on 27 January 2022).
- Zhang, X.; Walker, R.C.; Phizicky, E.M.; Mathews, D.H. Influence of Sequence and Covalent Modifications on Yeast TRNA Dynamics. J. Chem. Theory Comput. 2014, 10, 3473–3483. [Google Scholar] [CrossRef] [Green Version]
- Bavi, R.S.; Sambhare, S.B.; Sonawane, K.D. MD Simulation Studies to Investigate Iso-Energetic Conformational Behaviour of Modified Nucleosides m(2)G and m(2) 2G Present in TRNA. Comput. Struct. Biotechnol. J. 2013, 5, e201302015. [Google Scholar] [CrossRef] [Green Version]
- McCrate, N.E.; Varner, M.E.; Kim, K.I.; Nagan, M.C. Molecular Dynamics Simulations of Human TRNA Lys,3 UUU: The Role of Modified Bases in MRNA Recognition. Nucleic Acids Res. 2006, 34, 5361–5368. [Google Scholar] [CrossRef] [Green Version]
- Vangaveti, S.; Ranganathan, S.V.; Agris, P.F. Physical Chemistry of a Single TRNA-Modified Nucleoside Regulates Decoding of the Synonymous Lysine Wobble Codon and Affects Type 2 Diabetes. J. Phys. Chem. B 2022, 126, 1168–1177. [Google Scholar] [CrossRef]
- Sonawane, K.D.; Sambhare, S.B. The Influence of Hypermodified Nucleosides Lysidine and T6A to Recognize the AUA Codon Instead of AUG: A Molecular Dynamics Simulation Study. Int. Bio. 2015, 7, 1387–1395. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Van Voorhis, T. Nonlocal van Der Waals Density Functional: The Simpler the Better. J. Chem. Phys. 2010, 133, 244103. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a Package of Computer Programs for Applying Molecular Mechanics, Normal Mode Analysis, Molecular Dynamics and Free Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Yoluç, Y.; Ammann, G.; Barraud, P.; Jora, M.; Limbach, P.A.; Motorin, Y.; Marchand, V.; Tisné, C.; Borland, K.; Kellner, S. Instrumental Analysis of RNA Modifications. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 178–204. [Google Scholar] [CrossRef] [PubMed]
- Limbach, P.A.; Crain, P.F.; McCloskey, J.A. Molecular Mass Measurement of Intact Ribonucleic Acids via Electrospray Ionization Quadrupole Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1995, 6, 27–39. [Google Scholar] [CrossRef] [Green Version]
- McLuckey, S.A.; Van Berkel, G.J.; Glish, G.L. Tandem Mass Spectrometry of Small, Multiply Charged Oligonucleotides. J. Am. Soc. Mass Spectrom. 1992, 3, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Taucher, M.; Breuker, K. Top-down Mass Spectrometry for Sequencing of Larger (up to 61 Nt) RNA by CAD and EDD. J. Am. Soc. Mass Spectrom. 2010, 21, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Calderisi, G.; Glasner, H.; Breuker, K. Radical Transfer Dissociation for DE Novo Characterization of Modified Ribonucleic Acids by Mass Spectrometry. Angew. Chem. Weinheim Bergstr. Ger. 2020, 132, 4339–4343. [Google Scholar] [CrossRef] [Green Version]
- Peters-Clarke, T.M.; Quan, Q.; Brademan, D.R.; Hebert, A.S.; Westphall, M.S.; Coon, J.J. Ribonucleic Acid Sequence Characterization by Negative Electron Transfer Dissociation Mass Spectrometry. Anal. Chem. 2020, 92, 4436–4444. [Google Scholar] [CrossRef]
- Kowalak, J.A.; Pomerantz, S.C.; Crain, P.F.; McCloskey, J.A. A Novel Method for the Determination of Post-Transcriptional Modification in RNA by Mass Spectrometry. Nucleic Acids Res. 1993, 21, 4577–4585. [Google Scholar] [CrossRef] [Green Version]
- Thakur, P.; Estevez, M.; Lobue, P.A.; Limbach, P.A.; Addepalli, B. Improved RNA Modification Mapping of Cellular Non-Coding RNAs Using C- and U-Specific RNases. Analyst 2020, 145, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, N.; Kim, J.; Murgo, J.-R.; Kissai, M.; Ravichandran, K.; Miracco, E.J.; Presnyak, V.; Hua, S. Oligonucleotide Sequence Mapping of Large Therapeutic MRNAs via Parallel Ribonuclease Digestions and LC-MS/MS. Anal. Chem. 2019, 91, 8500–8506. [Google Scholar] [CrossRef] [PubMed]
- Solivio, B.; Yu, N.; Addepalli, B.; Limbach, P.A. Improving RNA Modification Mapping Sequence Coverage by LC-MS through a Nonspecific RNase U2-E49A Mutant. Anal. Chim. Acta 2018, 1036, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Crain, P.F. [42] Preparation and Enzymatic Hydrolysis of DNA and RNA for Mass Spectrometry. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1990; Volume 193, pp. 782–790. [Google Scholar]
- Cai, W.M.; Chionh, Y.H.; Hia, F.; Gu, C.; Kellner, S.; McBee, M.E.; Ng, C.S.; Pang, Y.L.J.; Prestwich, E.G.; Lim, K.S.; et al. A Platform for Discovery and Quantification of Modified Ribonucleosides in RNA: Application to Stress-Induced Reprogramming of TRNA Modifications. Methods Enzymol. 2015, 560, 29–71. [Google Scholar]
- Matuszewski, M.; Wojciechowski, J.; Miyauchi, K.; Gdaniec, Z.; Wolf, W.M.; Suzuki, T.; Sochacka, E. A Hydantoin Isoform of Cyclic N6-Threonylcarbamoyladenosine (Ct6A) Is Present in TRNAs. Nucleic Acids Res. 2017, 45, 2137–2149. [Google Scholar] [CrossRef] [Green Version]
- Jora, M.; Borland, K.; Abernathy, S.; Zhao, R.; Kelley, M.; Kellner, S.; Addepalli, B.; Limbach, P.A. Chemical Amination/Imination of Carbonothiolated Nucleosides during RNA Hydrolysis. Angew. Chem. Int. Engl. 2021, 60, 3961–3966. [Google Scholar] [CrossRef]
- Zhang, L.-S.; Liu, C.; Ma, H.; Dai, Q.; Sun, H.-L.; Luo, G.; Zhang, Z.; Zhang, L.; Hu, L.; Dong, X.; et al. Transcriptome-Wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian MRNA. Mol. Cell 2019, 74, 1304–1316.e8. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate Detection of M6A RNA Modifications in Native RNA Sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [Green Version]
- Amalric, A.; Bastide, A.; Attina, A.; Choquet, A.; Vialaret, J.; Lehmann, S.; David, A.; Hirtz, C. Quantifying RNA Modifications by Mass Spectrometry: A Novel Source of Biomarkers in Oncology. Crit. Rev. Clin. Lab. Sci. 2022, 59, 1–18. [Google Scholar] [CrossRef]
- Kaiser, S.; Byrne, S.R.; Ammann, G.; Asadi Atoi, P.; Borland, K.; Brecheisen, R.; DeMott, M.S.; Gehrke, T.; Hagelskamp, F.; Heiss, M.; et al. Strategies to Avoid Artifacts in Mass Spectrometry-Based Epitranscriptome Analyses. Angew. Chem. Int. Ed Engl. 2021, 60, 23885–23893. [Google Scholar] [CrossRef]
- Lauman, R.; Garcia, B.A. Unraveling the RNA Modification Code with Mass Spectrometry. Mol. Omics 2020, 16, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Porrini, M.; Rosu, F.; Rabin, C.; Darré, L.; Gómez, H.; Orozco, M.; Gabelica, V. Compaction of Duplex Nucleic Acids upon Native Electrospray Mass Spectrometry. ACS Cent Sci. 2017, 3, 454–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi-Ghanem, J.; Rabin, C.; Porrini, M.; Rosu, F.; Gabelica, V. Compaction of RNA Hairpins and Their Kissing Complexes in Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Wolff, P.; Ennifar, E. Native Electrospray Ionization Mass Spectrometry of RNA-Ligand Complexes. Methods Mol. Biol. 2020, 2113, 111–118. [Google Scholar]
- Schneeberger, E.-M.; Breuker, K. Native Top-down Mass Spectrometry of TAR RNA in Complexes with a Wild-type Tat Peptide for Binding Site Mapping. Angew. Chem. Weinheim Bergstr. Ger. 2017, 129, 1274–1278. [Google Scholar] [CrossRef]
- Hafner, M.; Katsantoni, M.; Köster, T.; Marks, J.; Mukherjee, J.; Staiger, D.; Ule, J.; Zavolan, M. CLIP and Complementary Methods. Nat. Rev. Methods Primers 2021, 1, 20. [Google Scholar] [CrossRef]
- Motorin, Y.; Helm, M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes 2019, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Sahadevan, S.; Pérez-Berlanga, M.; Polymenidou, M. Identification of RNA–RBP Interactions in Subcellular Compartments by CLIP-Seq. In The Integrated Stress Response: Methods and Protocols; Matějů, D., Chao, J.A., Eds.; Springer: New York, NY, USA, 2022; pp. 305–323. ISBN 9781071619759. [Google Scholar]
- Stork, C.; Zheng, S. Genome-Wide Profiling of RNA-Protein Interactions Using CLIP-Seq. Methods Mol. Biol. 2016, 1421, 137–151. [Google Scholar]
- Zhang, X.-Q.; Yang, J.-H. Discovering CircRNA-MicroRNA Interactions from CLIP-Seq Data. Methods Mol. Biol. 2018, 1724, 193–207. [Google Scholar]
- Vilfan, I.D.; Tsai, Y.-C.; Clark, T.A.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA Base Modification and Structural Rearrangement by Single-Molecule Real-Time Detection of Reverse Transcription. J. Nanobiotechnol. 2013, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications Using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Li, A.; Yu, N.J.; Nguyen, T.; Leach, R.W.; Wühr, M.; Kleiner, R.E. Activity-Based RNA-Modifying Enzyme Probing Reveals DUS3L-Mediated Dihydrouridylation. Nat. Chem. Biol. 2021, 17, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.J.; Turner, D.H. Optical Melting Measurements of Nucleic Acid Thermodynamics. Methods Enzymol. 2009, 468, 371–387. [Google Scholar]
- Mathews, D.H.; Turner, D.H.; Watson, R.M. RNA Secondary Structure Prediction. Curr. Protoc. Nucleic Acid Chem. 2016, 67, 11.2.1–11.2.19. [Google Scholar] [CrossRef] [Green Version]
- Dagneaux, C.; Liquier, J.; Taillandier, E. Sugar Conformations in DNA and RNA-DNA Triple Helixes Determined by FTIR Spectroscopy: Role of Backbone Composition. Biochemistry 1995, 34, 16618–16623. [Google Scholar] [CrossRef] [PubMed]
- Geinguenaud, F.; Militello, V.; Arluison, V. Application of FTIR Spectroscopy to Analyze RNA Structure. Methods Mol. Biol. 2020, 2113, 119–133. [Google Scholar]
- Tajmir-Riahi, H.A.; N’Soukpoe-Kossi, C.N.; Joly, D. Structural Analysis of Protein--DNA and Protein--RNA Interactions by FTIR, UV-Visible and CD Spectroscopic Methods. Spectroscopy 2009, 23, 81–101. [Google Scholar] [CrossRef]
- Desai, S.; Mishra, S.V.; Joshi, A.; Sarkar, D.; Hole, A.; Mishra, R.; Dutt, S.; Chilakapati, M.K.; Gupta, S.; Dutt, A. Raman Spectroscopy-Based Detection of RNA Viruses in Saliva: A Preliminary Report. J. Biophotonics 2020, 13, e202000189. [Google Scholar] [CrossRef]
- Gaston, H.B.H. Application of NIR Raman Spectroscopy to Probe the Flexibility of RNA Structure. Methods Mol. Biol. 2020, 2113, 149–164. [Google Scholar]
- Hobro, A.J.; Rouhi, M.; Blanch, E.W.; Conn, G.L. Raman and Raman Optical Activity (ROA) Analysis of RNA Structural Motifs in Domain I of the EMCV IRES. Nucleic Acids Res. 2007, 35, 1169–1177. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.; Johnson, J.E.; Thomas, G.J., Jr. Conformations, Interactions, and Thermostabilities of RNA and Proteins in Bean Pod Mottle Virus: Investigation of Solution and Crystal Structures by Laser Raman Spectroscopy. Biochemistry 1992, 31, 6673–6682. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.L.; Outeiral, C.; Dowd, S.E.; Doig, A.J.; Popelier, P.L.A.; Waltho, J.P.; Almond, A. Deconvolution of Conformational Exchange from Raman Spectra of Aqueous RNA Nucleosides. Commun. Chem. 2020, 3, 56. [Google Scholar] [CrossRef]
- Andrushchenko, V.; Wieser, H.; Bouř, P. RNA Structural Forms Studied by Vibrational Circular Dichroism: Ab initio Interpretation of the Spectra. J. Phys. Chem. B 2004, 108, 3899–3911. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, H.; Imahori, K. Circular Dichroism and Conformation of Natural and Synthetic Polynucleotides. J. Biochem. 1967, 61, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Sosnick, T.R.; Fang, X.; Shelton, V.M. [24] Application of Circular Dichroism to Study RNA Folding Transitions. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2000; Volume 317, pp. 393–409. [Google Scholar]
- Sugimoto, N.; Nakano, S.; Katoh, M.; Matsumura, A.; Nakamuta, H.; Ohmichi, T.; Yoneyama, M.; Sasaki, M. Thermodynamic Parameters to Predict Stability of RNA/DNA Hybrid Duplexes. Biochemistry 1995, 34, 11211–11216. [Google Scholar] [CrossRef]
- Vendeix, F.A.P.; Murphy, F.V., 4th; Cantara, W.A.; Leszczyńska, G.; Gustilo, E.M.; Sproat, B.; Malkiewicz, A.; Agris, P.F. Human TRNA(Lys3)(UUU) Is Pre-Structured by Natural Modifications for Cognate and Wobble Codon Binding through Keto-Enol Tautomerism. J. Mol. Biol. 2012, 416, 467–485. [Google Scholar] [CrossRef] [Green Version]
- Benvin, A.L.; Creeger, Y.; Fisher, G.W.; Ballou, B.; Waggoner, A.S.; Armitage, B.A. Fluorescent DNA Nanotags: Supramolecular Fluorescent Labels Based on Intercalating Dye Arrays Assembled on Nanostructured DNA Templates. J. Am. Chem. Soc. 2007, 129, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Bevilacqua, P.C.; Turner, D.H. Use of Fluorescence Spectroscopy to Elucidate RNA Folding Pathways. Curr. Protoc. Nucleic Acid Chem. 2002, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.R. Quantitation of DNA and RNA with Absorption and Fluorescence Spectroscopy. Curr. Protoc. Immunol. 2017, 116, A.3L.1–A.3L.14. [Google Scholar] [CrossRef]
- Liu, B.; Diamond, J.M.; Mathews, D.H.; Turner, D.H. Fluorescence Competition and Optical Melting Measurements of RNA Three-Way Multibranch Loops Provide a Revised Model for Thermodynamic Parameters. Biochemistry 2011, 50, 640–653. [Google Scholar] [CrossRef]
- Gaffarogullari, E.C.; Krause, A.; Balbo, J.; Herten, D.-P.; Jäschke, A. Microscale Thermophoresis Provides Insights into Mechanism and Thermodynamics of Ribozyme Catalysis. RNA Biol. 2013, 10, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.H.; Hilimire, T.A.; Sanders, A.M.; Schneekloth, J.S. Measuring RNA–Ligand Interactions with Microscale Thermophoresis. Biochemistry 2018, 57, 4638–4643. [Google Scholar] [CrossRef]
- Mrozowich, T.; MeierStephenson, V.; Patel, T.R. Microscale Thermophoresis: Warming up to a New Biomolecular Interaction Technique. Biochem. 2019, 41, 8–12. [Google Scholar] [CrossRef]
- Mao, S.; Haruehanroengra, P.; Ranganathan, S.V.; Shen, F.; Begley, T.J.; Sheng, J. Base Pairing and Functional Insights into N3-Methylcytidine (M3C) in RNA. ACS Chem. Biol. 2021, 16, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.T.; Fahrenbach, A.C.; Sheng, J.; Pian, J.; Szostak, J.W. Thermodynamic Insights into 2-Thiouridine-Enhanced RNA Hybridization. Nucleic Acids Res. 2015, 43, 7675–7687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agris, P.F.; Narendran, A.; Sarachan, K.; Väre, V.Y.P.; Eruysal, E. Chapter One—The Importance of Being Modified: The Role of RNA Modifications in Translational Fidelity. In The Enzymes; Chanfreau, G.F., Ed.; Academic Press: Cambridge, MA, USA, 2017; Volume 41, pp. 1–50. [Google Scholar]
- Davis, D.R.; Veltri, C.A.; Nielsen, L. An RNA Model System for Investigation of Pseudouridine Stabilization of the Codon-Anticodon Interaction in TRNALys, TRNAHis and TRNATyr. J. Biomol. Struct. Dyn. 1998, 15, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, I.; Kierzek, E.; Kierzek, R.; Schatz, G.C. Interplay of LNA and 2’-O-Methyl RNA in the Structure and Thermodynamics of RNA Hybrid Systems: A Molecular Dynamics Study Using the Revised AMBER Force Field and Comparison with Experimental Results. J. Phys. Chem. B 2014, 118, 14177–14187. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.S.; Ts’o, P.O.; von der Haar, F.; Sprinzl, M.; Cramer, F. NMR Study on the Methyl and Methylene Proton Resonances of TRNA Phe Yeast. Biochem. Biophys. Res. Commun. 1974, 59, 22–29. [Google Scholar] [CrossRef]
- Kastrup, R.V.; Schmidt, P.G. 1H NMR of Valine TRNA Modified Bases. Evidence for Multiple Conformations. Nucleic Acids Res. 1978, 5, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Liu, B.; Nussbaumer, F.; Rangadurai, A.; Kreutz, C.; Al-Hashimi, H.M. NMR Chemical Exchange Measurements Reveal That N6-Methyladenosine Slows RNA Annealing. J. Am. Chem. Soc. 2019, 141, 19988–19993. [Google Scholar] [CrossRef]
- Abou Assi, H.; Rangadurai, A.K.; Shi, H.; Liu, B.; Clay, M.C.; Erharter, K.; Kreutz, C.; Holley, C.L.; Al-Hashimi, H.M. 2′-O-Methylation Can Increase the Abundance and Lifetime of Alternative RNA Conformational States. Nucleic Acids Res. 2020, 48, 12365–12379. [Google Scholar] [CrossRef] [PubMed]
- Barraud, P.; Gato, A.; Heiss, M.; Catala, M.; Kellner, S.; Tisné, C. Time-Resolved NMR Monitoring of TRNA Maturation. Nat. Commun. 2019, 10, 3373. [Google Scholar] [CrossRef] [Green Version]
- Fürtig, B.; Richter, C.; Wöhnert, J.; Schwalbe, H. NMR Spectroscopy of RNA. Chembiochem 2003, 4, 936–962. [Google Scholar] [CrossRef] [PubMed]
- Aguion, P.I.; Marchanka, A. Strategies for RNA Resonance Assignment by 13C/15N- and 1H-Detected Solid-State NMR Spectroscopy. Front Mol Biosci 2021, 8, 743181. [Google Scholar] [CrossRef]
- Pujari, N.; Saundh, S.L.; Acquah, F.A.; Mooers, B.H.M.; Ferré-D’Amaré, A.R.; Leung, A.K.-W. Engineering Crystal Packing in RNA Structures I: Past and Future Strategies for Engineering RNA Packing in Crystals. Crystals 2021, 11, 952. [Google Scholar] [CrossRef]
- Edwards, A.L.; Garst, A.D.; Batey, R.T. Determining Structures of RNA Aptamers and Riboswitches by X-Ray Crystallography. Methods Mol. Biol. 2009, 535, 135–163. [Google Scholar] [PubMed] [Green Version]
- Reyes, F.E.; Garst, A.D.; Batey, R.T. Strategies in RNA Crystallography. Methods Enzymol. 2009, 469, 119–139. [Google Scholar]
- Keel, A.Y.; Rambo, R.P.; Batey, R.T.; Kieft, J.S. A General Strategy to Solve the Phase Problem in RNA Crystallography. Structure 2007, 15, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Kappel, K.; Zhang, K.; Su, Z.; Watkins, A.M.; Kladwang, W.; Li, S.; Pintilie, G.; Topkar, V.V.; Rangan, R.; Zheludev, I.N.; et al. Accelerated Cryo-EM-Guided Determination of Three-Dimensional RNA-Only Structures. Nat. Methods 2020, 17, 699–707. [Google Scholar] [CrossRef]
- Wang, H.-W.; Wang, J.-W. How Cryo-Electron Microscopy and X-Ray Crystallography Complement Each Other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Kretsch, R.; Das, R.; Chiu, W. IUCr Interpretation of RNA Cryo-EM Maps of Various Resolutions. Acta Crystallogr. Sect. A Found. Adv. 2021, 77, a217. [Google Scholar] [CrossRef]
- Das, R. RNA Structure: A Renaissance Begins? Nat. Methods 2021, 18, 439. [Google Scholar] [CrossRef] [PubMed]
- Hopfinger, M.C.; Kirkpatrick, C.C.; Znosko, B.M. Predictions and Analyses of RNA Nearest Neighbor Parameters for Modified Nucleotides. Nucleic Acids Res. 2020, 48, 8901–8913. [Google Scholar] [CrossRef]
- Tan, D.; Piana, S.; Dirks, R.M.; Shaw, D.E. RNA Force Field with Accuracy Comparable to State-of-the-Art Protein Force Fields. Proc. Natl. Acad. Sci. USA 2018, 115, E1346–E1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergonzo, C.; Henriksen, N.M.; Roe, D.R.; Cheatham, T.E., 3rd. Highly Sampled Tetranucleotide and Tetraloop Motifs Enable Evaluation of Common RNA Force Fields. RNA 2015, 21, 1578–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE 2012, 7, e32131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, I.; Stern, H.A.; Tubbs, J.D.; Kennedy, S.D.; Turner, D.H. Benchmarking AMBER Force Fields for RNA: Comparisons to NMR Spectra for Single-Stranded r(GACC) Are Improved by Revised χ Torsions. J. Phys. Chem. B 2011, 115, 9261–9270. [Google Scholar] [CrossRef]
- Condon, D.E.; Kennedy, S.D.; Mort, B.C.; Kierzek, R.; Yildirim, I.; Turner, D.H. Stacking in RNA: NMR of Four Tetramers Benchmark Molecular Dynamics. J. Chem. Theory Comput. 2015, 11, 2729–2742. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Methods | Advantages | Disadvantages | Computational Information |
|---|---|---|---|
| Mass Spectrometry | Native solvent conditions Attomolar concentrations can be used | No 3D insight Sample is not recoverable Size limitations Gas phase experiments | Chemical ID Sequence position |
| Sequencing Techniques | Single nucleotide resolution Population or single molecule-based methods available | Mediocre accuracy and precision in detection | Sequence position |
| UV Optical Experiments | Micromolar concentrations can be used Fast experimentation Thermodynamics insight | Two state dependent No insight beyond helical stability | Melting temperature Helical stabilityChanges in free energy, enthalpy, and entropy |
| NMR | Native conditions Sensitive to structure fluctuations | Size limitation Lengthy data interpretation 3D molecule resolution difficult to attain | Distance restraints Nucleotide/RNA 3D orientation Secondary structure (base pairing/non-paired) |
| X-ray Crystallography | 3D structure can be determined | RNAs are hard to crystallize Non-native conditions Requires homogeneous crystals | 3D coordinates and orientation of RNA molecule |
| Cryo-EM | Heterogeneous populations detectable Crystals not necessary Native conditions | Data collection, analysis, and troubleshooting is lengthy and complex | 3D coordinates and orientation of RNA molecule Tertiary contacts detectable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Esposito, R.J.; Myers, C.A.; Chen, A.A.; Vangaveti, S. Challenges with Simulating Modified RNA: Insights into Role and Reciprocity of Experimental and Computational Approaches. Genes 2022, 13, 540. https://doi.org/10.3390/genes13030540
D’Esposito RJ, Myers CA, Chen AA, Vangaveti S. Challenges with Simulating Modified RNA: Insights into Role and Reciprocity of Experimental and Computational Approaches. Genes. 2022; 13(3):540. https://doi.org/10.3390/genes13030540
Chicago/Turabian StyleD’Esposito, Rebecca J., Christopher A. Myers, Alan A. Chen, and Sweta Vangaveti. 2022. "Challenges with Simulating Modified RNA: Insights into Role and Reciprocity of Experimental and Computational Approaches" Genes 13, no. 3: 540. https://doi.org/10.3390/genes13030540
APA StyleD’Esposito, R. J., Myers, C. A., Chen, A. A., & Vangaveti, S. (2022). Challenges with Simulating Modified RNA: Insights into Role and Reciprocity of Experimental and Computational Approaches. Genes, 13(3), 540. https://doi.org/10.3390/genes13030540