
Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow



Abstract
:
1. Introduction
2. Materials and Methods
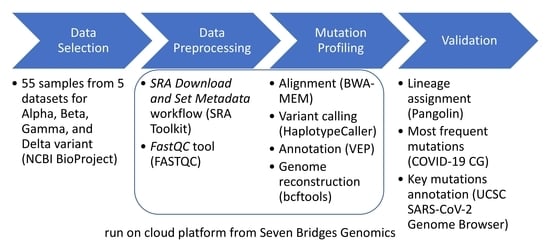
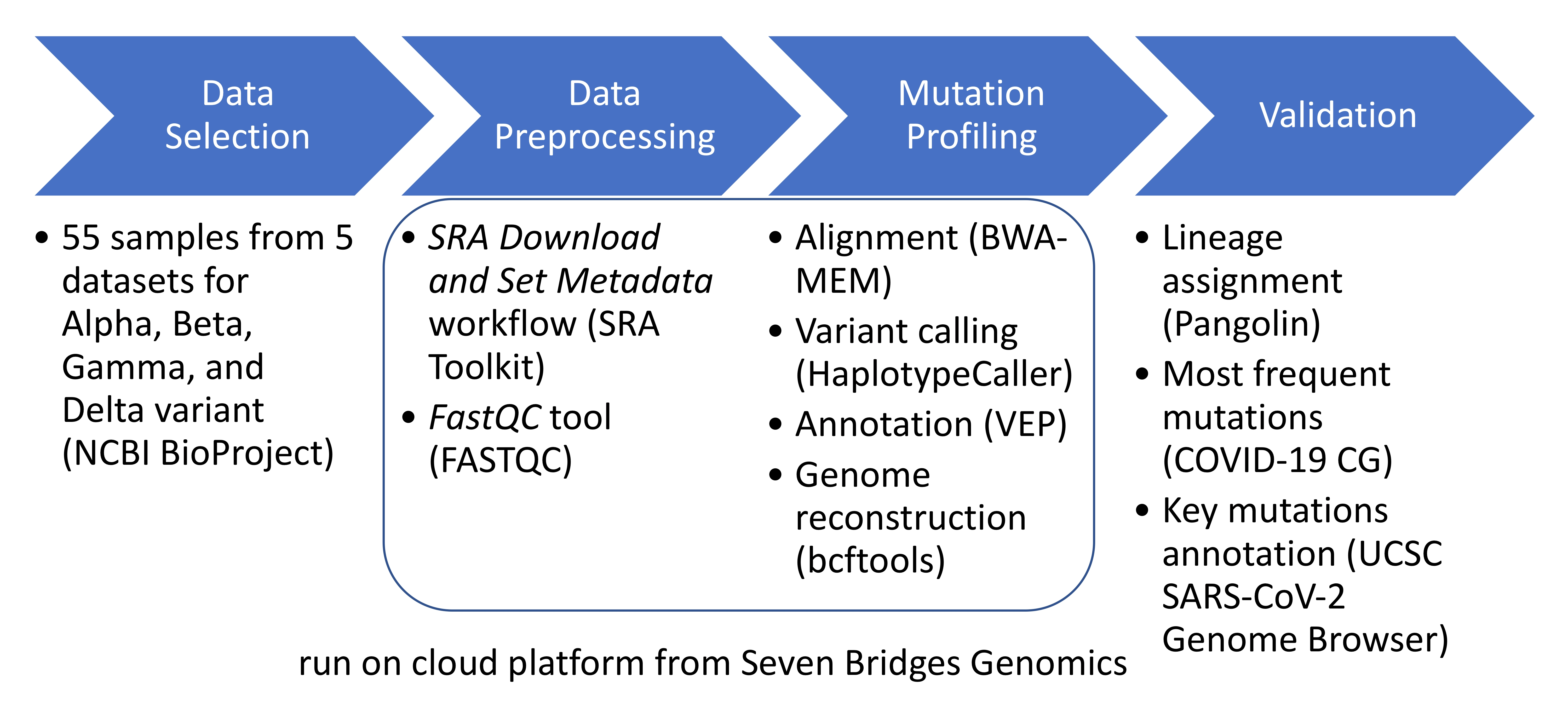
2.1. Data Selection
2.2. Data Preprocessing
2.3. Mutation Profiling
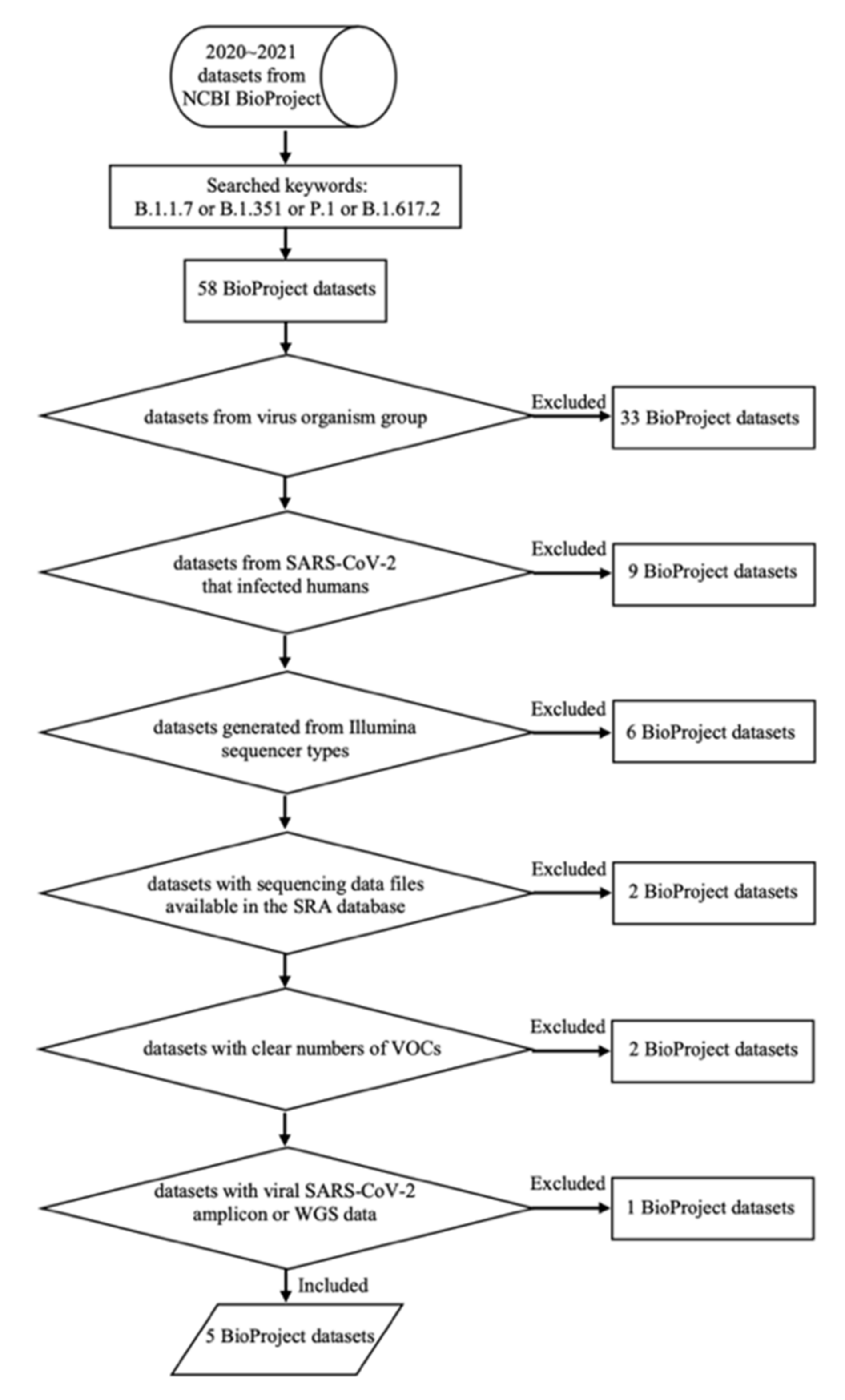
2.3.1. Cloud Workflow Design
2.3.2. Validation
3. Results
3.1. Cloud Workflow Performance
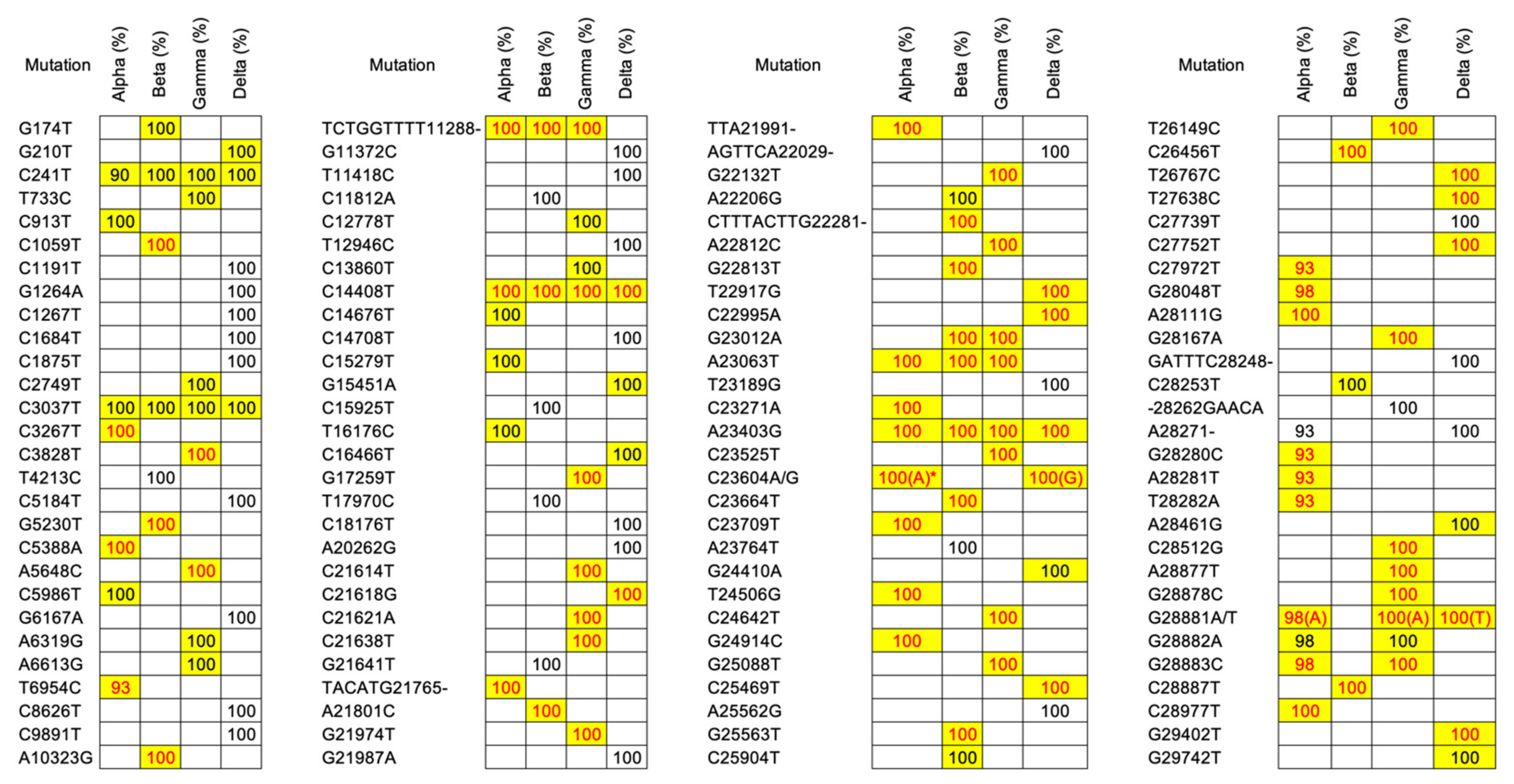
3.2. Mutation Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naming the Coronavirus Disease (COVID-19) and the Virus that Causes It. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 31 December 2021).
- Mahase, E. COVID-19: WHO declares pandemic because of “alarming levels” of spread, severity, and inaction. BMJ 2020, 368, m1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- WHO Announces Simple, Easy-to-Say Labels for SARS-CoV-2 Variants of Interest and Concern. Available online: https://www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern (accessed on 31 December 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- O’Toole, A.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; COVID, T.; UK, G. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 with grinch. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 31 December 2021).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Gong, Z.; Zhu, J.W.; Li, C.P.; Jiang, S.; Ma, L.N.; Tang, B.X.; Zou, D.; Chen, M.L.; Sun, Y.B.; Song, S.H.; et al. An online coronavirus analysis platform from the National Genomics Data Center. Zool. Res. 2020, 41, 705–708. [Google Scholar] [CrossRef]
- Strozzi, F.; Janssen, R.; Wurmus, R.; Crusoe, M.R.; Githinji, G.; di Tommaso, P.; Belhachemi, D.; Moller, S.; Smant, G.; de Ligt, J.; et al. Scalable workflows and reproducible data analysis for genomics. In Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1910, pp. 723–745. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.G.; Hsiao, S.H.; Lee, Y.G. Orchestrating an Optimized Next-Generation Sequencing-Based Cloud Workflow for Robust Viral Identification during Pandemics. Biology 2021, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Holzer, M. Porecov-An Easy to Use, Fast, and Robust Workflow for SARS-CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2021, 12, 711437. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Workflow systems turn raw data into scientific knowledge. Nature 2019, 573, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief. Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; van Kerkhove, M.D.; Jombart, T.; et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eurosurveillance 2021, 26, 2100509. [Google Scholar] [CrossRef]
- Barrett, T.; Clark, K.; Gevorgyan, R.; Gorelenkov, V.; Gribov, E.; Karsch-Mizrachi, I.; Kimelman, M.; Pruitt, K.D.; Resenchuk, S.; Tatusova, T.; et al. BioProject and BioSample databases at NCBI: Facilitating capture and organization of metadata. Nucleic Acids Res. 2012, 40, D57–D63. [Google Scholar] [CrossRef]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- Lau, J.W.; Lehnert, E.; Sethi, A.; Malhotra, R.; Kaushik, G.; Onder, Z.; Groves-Kirkby, N.; Mihajlovic, A.; DiGiovanna, J.; Srdic, M.; et al. The Cancer Genomics Cloud: Collaborative, Reproducible, and Democratized-A New Paradigm in Large-Scale Computational Research. Cancer Res. 2017, 77, e3–e6. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, G.; Ivkovic, S.; Simonovic, J.; Tijanic, N.; Davis-Dusenbery, B.; Kural, D. Rabix: An open-source workflow executor supporting recomputability and interoperability of workflow descriptions. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2017; Volume 22, pp. 154–165. [Google Scholar] [CrossRef] [Green Version]
- Amstutz, P.; Crusoe, M.R.; Tijanić, N.; Chapman, B.; Chilton, J.; Heuer, M.; Kartashov, A.; Leehr, D.; Ménager, H.; Nedeljkovich, M.; et al. Common Workflow Language, v1.0. Figshare 2016. [Google Scholar] [CrossRef]
- Team, S.T.D. The NCBI SRA Toolkit. Available online: https://github.com/ncbi/sra-tools (accessed on 31 December 2021).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 31 December 2021).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef]
- Tischler, G.; Leonard, S. biobambam: Tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 2014, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Chen, A.T.; Altschuler, K.; Zhan, S.H.; Chan, Y.A.; Deverman, B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. eLife 2021, 10, e63409. [Google Scholar] [CrossRef]
- Fernandes, J.D.; Hinrichs, A.S.; Clawson, H.; Gonzalez, J.N.; Lee, B.T.; Nassar, L.R.; Raney, B.J.; Rosenbloom, K.R.; Nerli, S.; Rao, A.A.; et al. The UCSC SARS-CoV-2 Genome Browser. Nat. Genet. 2020, 52, 991–998. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Al-Mulla, F.; Wei, D.Q.; Abubaker, J. Remdesivir MD Simulations Suggest a More Favourable Binding to SARS-CoV-2 RNA Dependent RNA Polymerase Mutant P323L Than Wild-Type. Biomolecules 2021, 11, 919. [Google Scholar] [CrossRef]
- Yang, H.C.; Chen, C.H.; Wang, J.H.; Liao, H.C.; Yang, C.T.; Chen, C.W.; Lin, Y.C.; Kao, C.H.; Lu, M.J.; Liao, J.C. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Lin, L.; Liu, Y.; Tang, X.; He, D. The Disease Severity and Clinical Outcomes of the SARS-CoV-2 Variants of Concern. Front. Public Health 2021, 9, 775224. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, C.A. Could mutations of SARS-CoV-2 suppress diagnostic detection? Nat. Biotechnol. 2021, 39, 274–275. [Google Scholar] [CrossRef] [PubMed]
- World Economic Situation and Prospects. Available online: https://www.un.org/development/desa/dpad/wp-content/uploads/sites/45/WESP2020_Annex.pdf (accessed on 31 December 2021).
- Wratten, L.; Wilm, A.; Goke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat. Methods 2021, 18, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, M.; Zhang, N.; Harbers, L.; Milia, M.G.; Brossa, S.; Huong Nguyen, T.T.; Cerutti, F.; Berrino, E.; Sapino, A.; Bienko, M.; et al. COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance. Nat. Commun. 2021, 12, 3903. [Google Scholar] [CrossRef] [PubMed]
- Posada-Cespedes, S.; Seifert, D.; Topolsky, I.; Jablonski, K.P.; Metzner, K.J.; Beerenwinkel, N. V-pipe: A computational pipeline for assessing viral genetic diversity from high-throughput data. Bioinformatics 2021, 37, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Hodcroft, E.B.; de Maio, N.; Lanfear, R.; MacCannell, D.R.; Minh, B.Q.; Schmidt, H.A.; Stamatakis, A.; Goldman, N.; Dessimoz, C. Want to track pandemic variants faster? Fix the bioinformatics bottleneck. Nature 2021, 591, 30–33. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant. | Lineage * | Alias | Place of Origin | Time of Origin | Type | Designation |
|---|---|---|---|---|---|---|
| α (Alpha) | B.1.1.7 | - | United Kingdom | September 2020 | VOC | 18 December 2020 |
| β (Beta) | B.1.351 | - | South Africa | May 2020 | VOC | 18 December 2020 |
| γ (Gamma) | P.1 | B.1.1.28.1 | Brazil | November 2020 | VOC | 11 January 2021 |
| δ (Delta) | B.1.617.2 | - | India | October 2020 | VOC | 11 May 2021 |
| ε (Epsilon) | B.1.427/B.1.429 | - | United States | March 2020 | VOI | 5 March 2021 |
| ζ (Zeta) | P.2 | B.1.1.28.2 | Brazil | April 2020 | VOI | 17 March 2021 |
| η (Eta) | B.1.525 | - | multiple countries | December 2020 | VOI | 17 March 2021 |
| θ (Theta) | P.3 | B.1.1.28.3 | Philippines | January 2021 | VOI | 24 March 2021 |
| ι (Iota) | B.1.526 | - | United States | November 2020 | VOI | 24 March 2021 |
| κ (Kappa) | B.1.617.1 | - | India | October 2020 | VOI | 4 April 2021 |
| λ (Lambda) | C.37 | B.1.1.1.37 | Peru | December 2020 | VOI | 14 June 2021 |
| μ (Mu) | B.1.621 | - | Colombia | January 2021 | VOI | 30 August 2021 |
| ο (Omicron) | B.1.1.529 | - | multiple countries | November 2021 | VOC | 26 November 2021 |
| Data Set | No. of Samples | No. of Reads | Sequencer Type | Sequencing Strategy | |||
|---|---|---|---|---|---|---|---|
| Alpha | Beta | Gamma | Delta | ||||
| PRJNA704235 | - | - | 3 | - | 1,483,054 | MiniSeq | WGS |
| PRJNA708134 | 39 | - | - | - | 16,060,411 | NovaSeq | amplicon |
| PRJNA726840 | 3 | 8 | - | - | 2,801,089 | iSeq | WGS |
| PRJNA726871 | - | - | - | 1 | 128,048 | MiSeq | amplicon |
| PRJNA733209 | - | - | 1 | - | 2,415,993 | MiniSeq | WGS |
| Total | 42 | 8 | 4 | 1 | 22,888,595 | - | - |
| Step | Name | Instance 1 | Time 2 | Cost 3 |
|---|---|---|---|---|
| Data preprocessing | SRA Download and Set Metadata workflow | c4.8xlarge a | 7 | 0.09 |
| FastQC tool | c4.2xlarge b | 6 | 0.04 | |
| Mutation Profiling | Our cloud workflow * | c4.2xlarge | 7 | 2.09 |
| Total | 20 | 2.22 | ||
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C3267T | SNV | moderate | ORF1ab | aCt3002aTt | T1001I | nsp3 |
| C5388A | SNV | moderate | ORF1ab | gCt5123gAt | A1708D | nsp3 |
| T6954C | SNV | moderate | ORF1ab | aTa6689aCa | I2230T | nsp3 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| TACATG21765- | deletion | moderate | S | aTACATGtc 203:208atc | IHV68:70I | S |
| TTA21991- | deletion | moderate | S | gtTTAt 429-431gtt | VY143:144V | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| C23271A | SNV | moderate | S | gCt1709gAt | A570D | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23604A | SNV | moderate | S | cCt2042cAt | P681H | S |
| C23709T | SNV | moderate | S | aCa2147aTa | T716I | S |
| T24506G | SNV | moderate | S | Tca2944Gca | S982A | S |
| G24914C | SNV | moderate | S | Gac3352Cac | D1118H | S |
| C27972T | SNV | high | ORF8 | Caa79Taa | Q27stop | ORF8 |
| G28048T | SNV | moderate | ORF8 | aGa155aTa | R52I | ORF8 |
| A28111G | SNV | moderate | ORF8 | tAc218tGc | Y73C | ORF8 |
| G28280C | SNV | moderate | N | Gat7Cat | D3H | N |
| A28281T | SNV | moderate | N | gAt8gTt | D3V | N |
| T28282A | SNV | moderate | N | gaT9gaA | D3E | N |
| G28881A | SNV | moderate | N | aGg608aAg | R203K | N |
| G28883C | SNV | moderate | N | Gga610Cga | G204R | N |
| C28977T | SNV | moderate | N | tCt704tTt | S235F | N |
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C1059T | SNV | moderate | ORF1ab | aCc794aTc | T265I | nsp2 |
| G5230T | SNV | moderate | ORF1ab | aaG4965aaT | K1655N | nsp3 |
| A10323G | SNV | moderate | ORF1ab | aAg10058aGg | K3353R | nsp5 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| A21801C | SNV | moderate | S | gAt239gCt | D80A | S |
| CTTTACTTG22281- | deletion | moderate | S | aCTTTACTTGct719-727act | TLLA240:243T | S |
| G22813T | SNV | moderate | S | aaG1251aaT | K417N | S |
| G23012A | SNV | moderate | S | Gaa1450Aaa | E484K | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23664T | SNV | moderate | S | gCa2102gTa | A701V | S |
| G25563T | SNV | moderate | ORF3a | caG171caT | Q57H | ORF3a |
| C26456T | SNV | moderate | E | cCt212cTt | P71L | E |
| C28887T | SNV | moderate | N | aCt614aTt | T205I | N |
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C3828T | SNV | moderate | ORF1ab | tCa3563tTa | S1188L | nsp3 |
| A5648C | SNV | moderate | ORF1ab | Aaa5383Caa | K1795Q | nsp3 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| G17259T | SNV | moderate | ORF1ab | gaG16995gaT | E5665D | nsp13 |
| C21614T | SNV | moderate | S | Ctt52Ttt | L18F | S |
| C21621A | SNV | moderate | S | aCc59aAc | T20N | S |
| C21638T | SNV | moderate | S | Cct76Tct | P26S | S |
| G21974T | SNV | moderate | S | Gat412Tat | D138Y | S |
| G22132T | SNV | moderate | S | agG570agT | R190S | S |
| A22812C | SNV | moderate | S | aAg1250aCg | K417T | S |
| G23012A | SNV | moderate | S | Gaa1450Aaa | E484K | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23525T | SNV | moderate | S | Cat1963Tat | H655Y | S |
| C24642T | SNV | moderate | S | aCt3080aTt | T1027I | S |
| G25088T | SNV | moderate | S | Gtt3526Ttt | V1176F | S |
| T26149C | SNV | moderate | ORF3a | Tcc757Ccc | S253P | ORF3a |
| G28167A | SNV | moderate | ORF8 | Gaa274Aaa | E92K | ORF8 |
| C28512G | SNV | moderate | N | cCa239cGa | P80R | N |
| A28877T | SNV | moderate | N | Agt604Tgt | S202C | N |
| G28878C | SNV | moderate | N | aGt605aCt | S202T | N |
| G28881A | SNV | moderate | N | aGg608aAg | R203K | N |
| G28883C | SNV | moderate | N | Gga610Cga | G204R | N |
| Mutation. | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| C21618G | SNV | moderate | S | aCa56aGa | T19R | S |
| T22917G | SNV | moderate | S | cTg1355cGg | L452R | S |
| C22995A | SNV | moderate | S | aCa1433aAa | T478K | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23604G | SNV | moderate | S | cCt2042cGt | P681R | S |
| C25469T | SNV | moderate | ORF3a | tCa77tTa | S26L | ORF3a |
| T26767C | SNV | moderate | M | aTc245aCc | I82T | M |
| T27638C | SNV | moderate | ORF7a | gTt245gCt | V82A | ORF7a |
| C27752T | SNV | moderate | ORF7a | aCa359aTa | T120I | ORF7a |
| G28881T | SNV | moderate | N | aGg608aTg | R203M | N |
| G29402T | SNV | moderate | N | Gat1129Tat | D377Y | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, H.G.-M.; Hsiao, S.-H.; Fann, Y.C.; Lee, Y.-C.G. Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes 2022, 13, 686. https://doi.org/10.3390/genes13040686
Lim HG-M, Hsiao S-H, Fann YC, Lee Y-CG. Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes. 2022; 13(4):686. https://doi.org/10.3390/genes13040686
Chicago/Turabian StyleLim, Hendrick Gao-Min, Shih-Hsin Hsiao, Yang C. Fann, and Yuan-Chii Gladys Lee. 2022. "Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow" Genes 13, no. 4: 686. https://doi.org/10.3390/genes13040686
APA StyleLim, H. G. -M., Hsiao, S. -H., Fann, Y. C., & Lee, Y. -C. G. (2022). Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes, 13(4), 686. https://doi.org/10.3390/genes13040686