
Gene-Based Association Tests Using New Polygenic Risk Scores and Incorporating Gene Expression Data

Abstract
:1. Introduction
2. Methods
2.1. TWAS
2.2. Newly Developed LD-Adjusted PRSs
2.3. Association Test Leveraging Both Gene Expression Measurements and PRSs
3. Comparison of Methods
4. Simulations
5. Simulation Results
5.1. Type I Error Rates
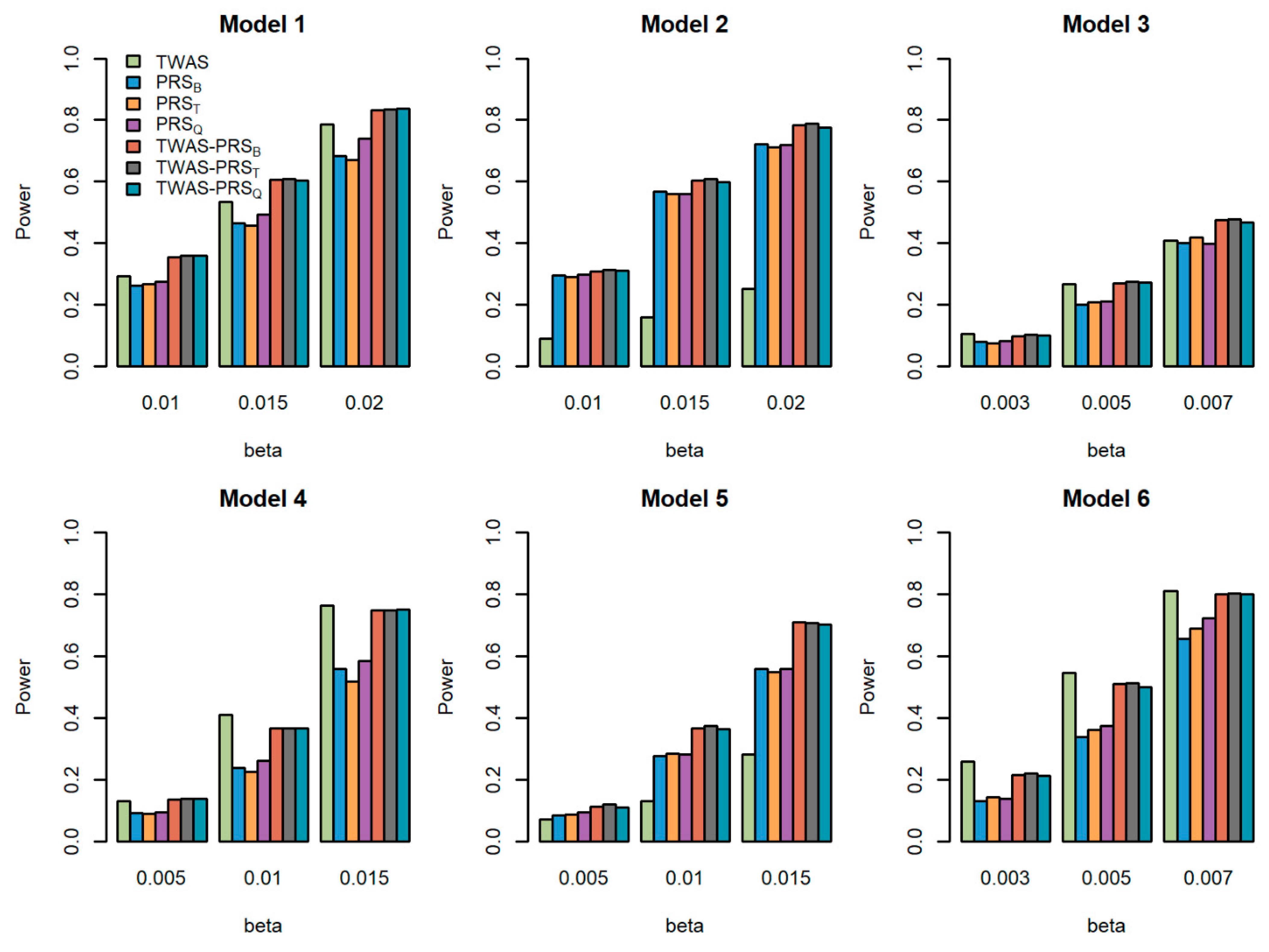
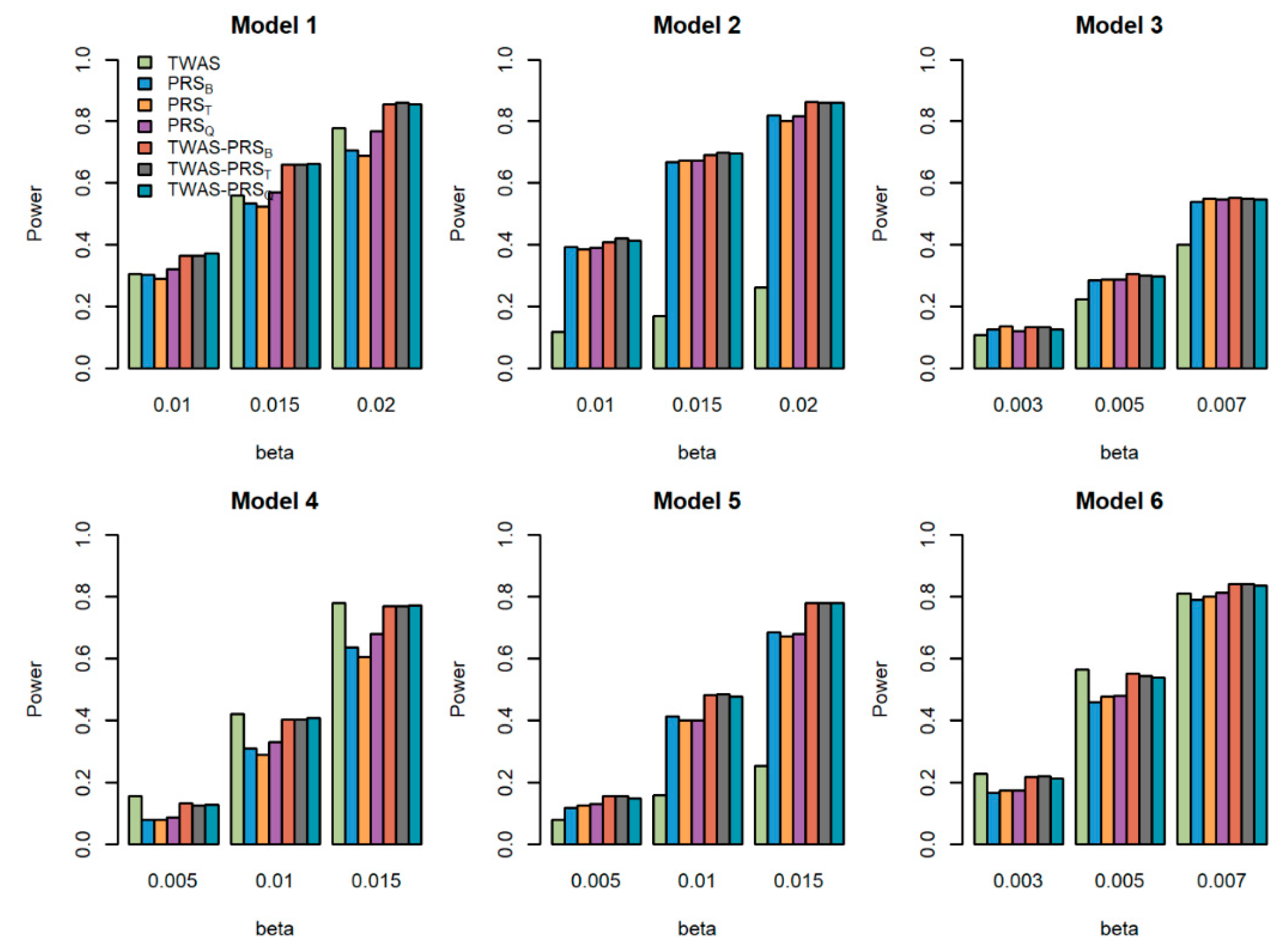
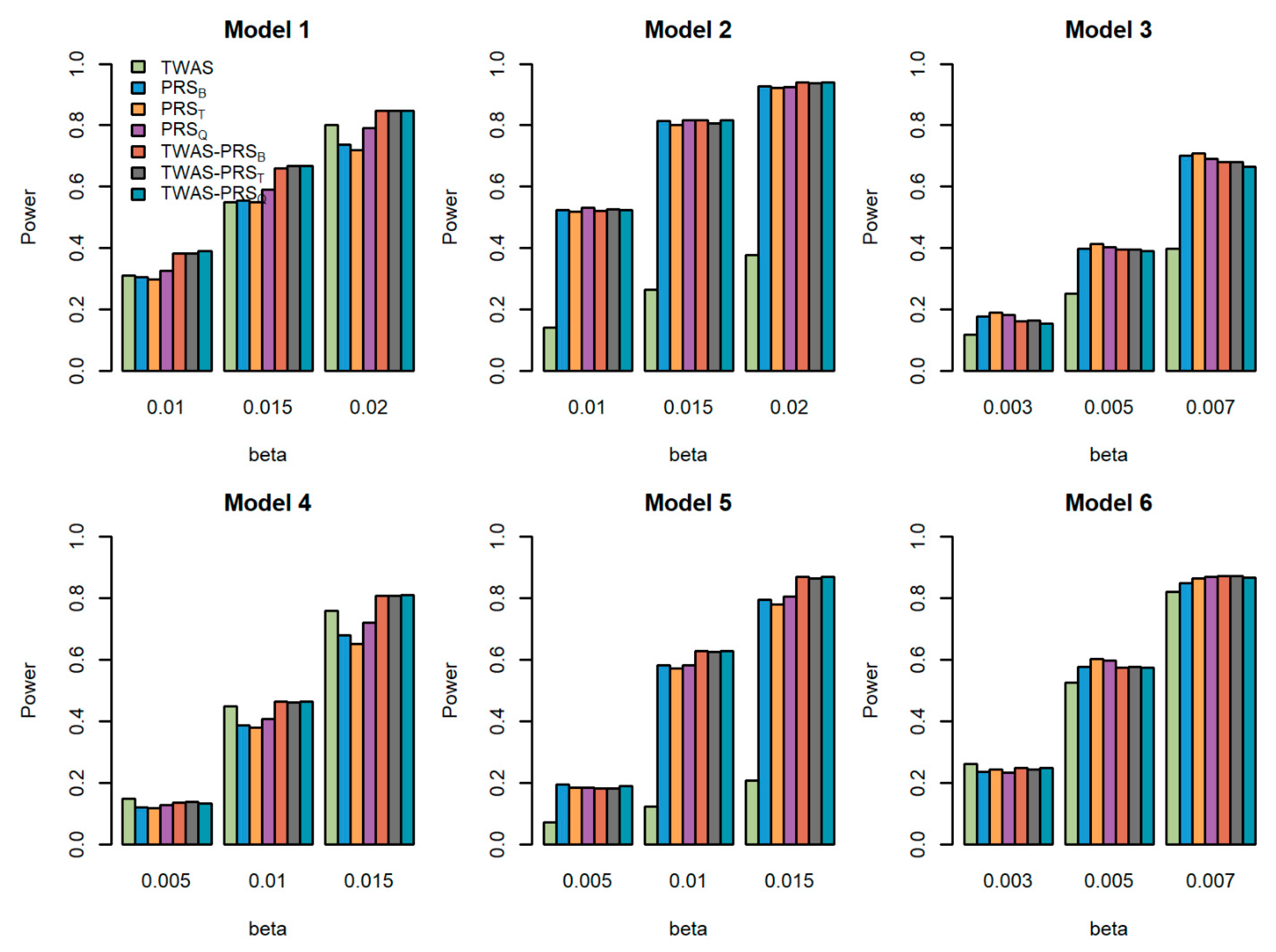
5.2. Powers
6. Application to UK Biobank Data
6.1. UK Biobank Data
6.2. Results
7. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eichler, E.E.; Flint, J.; Gibson, G.; Kong, A.; Leal, S.M.; Moore, J.H.; Nadeau, J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010, 11, 446–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visscher, P.M.; Hill, W.G.; Wray, N.R. Heritability in the genomics era—Concepts and misconceptions. Nat. Rev. Genet. 2008, 9, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, I.S. Common polygenic variation contributes to risk of schizophrenia that overlaps with bipolar disorder. Nature 2009, 460, 748. [Google Scholar]
- Torkamani, A.; Wineinger, N.E.; Topol, E.J. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018, 19, 581–590. [Google Scholar] [CrossRef]
- Ripatti, S.; Tikkanen, E.; Orho-Melander, M.; Havulinna, A.S.; Silander, K.; Sharma, A.; Guiducci, C.; Perola, M.; Jula, A.; Sinisalo, J.; et al. A multilocus genetic risk score for coronary heart disease: Case-control and prospective cohort analyses. Lancet 2010, 376, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.M.; Lawlor, D.A.; Harbord, R.M.; Sheehan, N.A.; Tobias, J.H.; Timpson, N.J.; Smith, G.D.; Sterne, J. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat. Methods Med. Res. 2011, 21, 223–242. [Google Scholar] [CrossRef] [Green Version]
- Gamazon, E.R.; GTEx Consortium; Wheeler, H.E.; Shah, K.P.; Mozaffari, S.V.; Aquino-Michaels, K.; Carroll, R.J.; Eyler, A.E.; Denny, J.C.; Nicolae, D.L.; et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015, 47, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.; Schmidt, K.M.; Sims, R.; O’Donovan, M.C.; Williams, J.; Holmans, P.; Escott-Price, V.; GERAD Consortium. POLARIS: Polygenic LD-adjusted risk score approach for set-based analysis of GWAS data. Genet. Epidemiol. 2018, 42, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wu, C.; Wei, P.; Pan, W. A Powerful Framework for Integrating eQTL and GWAS Summary Data. Genetics 2017, 207, 893–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veturi, Y.; Ritchie, M.D. (Eds.) How powerful are summary-based methods for identifying expression-trait associations under different genetic architectures? In Proceedings of the Pacific Symposium 2018, Big Island, HI, USA, 3–7 January 2018. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Sha, Q.; Zhang, Z.; Zhang, S. An improved score test for genetic association studies. Genet. Epidemiol. 2011, 35, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P. Tutorial: A guide to performing polygenic risk score analyses. Nat. Protoc. 2020, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Wheeler, B.; Sampson, J.N.; Hartge, P.; Chanock, S.J.; Park, J.-H. Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat. Genet. 2013, 45, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Sha, Q.; Wang, X.; Wang, X.; Zhang, S. Detecting association of rare and common variants by testing an optimally weighted combination of variants. Genet. Epidemiol. 2012, 36, 561–571. [Google Scholar] [CrossRef]
- Regan, E.A.; Hokanson, J.E.; Murphy, J.R.; Make, B.; Lynch, D.A.; Beaty, T.H.; Curran-Everett, D.; Silverman, E.K.; Crapo, J.D. Genetic epidemiology of COPD (COPDGene) study design. COPD J. Chronic Obstr. Pulm. Dis. 2011, 7, 32–43. [Google Scholar] [CrossRef]
- Scheet, P.; Stephens, M. A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Wang, Z.; Sha, Q.; Zhang, S. An Adaptive Fisher’s Combination Method for Joint Analysis of Multiple Phenotypes in Association Studies. Sci. Rep. 2016, 6, srep34323. [Google Scholar] [CrossRef]
- Biobank, U. UK Biobank: Protocol for a large-scale prospective epidemiological resource. Accessed May 2007, 7, 1–112. [Google Scholar]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Jia, Q.; Jahani, P.S.; Hurrell, B.P.; Pan, C.; Huang, P.; Gukasyan, J.; Woodward, N.C.; Eskin, E.; Gilliland, F.D.; et al. Genome-wide analysis highlights contribution of immune system pathways to the genetic architecture of asthma. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerez, F.; Plaza, V.; Tarrega, J.; Casan, P.; Rodriguez, J. Thyroid function and difficult to manage asthma. Arch. Bronconeumol. 1998, 34, 429–432. [Google Scholar] [CrossRef]
- Dong, Z.; Ma, Y.; Zhou, H.; Shi, L.; Ye, G.; Yang, L.; Liu, P.; Zhou, L. Integrated genomics analysis highlights important SNPs and genes implicated in moderate-to-severe asthma based on GWAS and eQTL datasets. BMC Pulm. Med. 2020, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Fuller, C.K.; Song, Y.; Meng, Q.; Zhang, B.; Yang, X.; Li, H. Sherlock: Detecting Gene-Disease Associations by Matching Patterns of Expression QTL and GWAS. Am. J. Hum. Genet. 2013, 92, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Valette, K.; Li, Z.; Bon-Baret, V.; Chignon, A.; Bérubé, J.-C.; Eslami, A.; Lamothe, J.; Gaudreault, N.; Joubert, P.; Obeidat, M.; et al. Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK Biobank. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | TWAS | PRSB | PRST | PRSQ | TWAS-PRSB | TWAS-PRST | TWAS-PRSQ | |
|---|---|---|---|---|---|---|---|---|
| 5000 | 1 | 0.044 | 0.056 | 0.062 | 0.057 | 0.056 | 0.057 | 0.058 |
| 2 | 0.048 | 0.051 | 0.048 | 0.050 | 0.063 | 0.061 | 0.063 | |
| 3 | 0.046 | 0.042 | 0.045 | 0.045 | 0.049 | 0.051 | 0.050 | |
| 10,000 | 1 | 0.044 | 0.055 | 0.057 | 0.051 | 0.060 | 0.063 | 0.058 |
| 2 | 0.054 | 0.046 | 0.047 | 0.049 | 0.052 | 0.047 | 0.047 | |
| 3 | 0.050 | 0.052 | 0.054 | 0.056 | 0.060 | 0.057 | 0.046 | |
| 20,000 | 1 | 0.043 | 0.049 | 0.047 | 0.047 | 0.054 | 0.051 | 0.055 |
| 2 | 0.039 | 0.040 | 0.040 | 0.041 | 0.043 | 0.044 | 0.047 | |
| 3 | 0.040 | 0.042 | 0.039 | 0.047 | 0.040 | 0.042 | 0.042 |
| Setting | TWAS | PRSB | PRST | PRSQ | TWAS-PRSB | TWAS-PRST | TWAS-PRSQ |
|---|---|---|---|---|---|---|---|
| 47 (28) | 190 (98) | 198 (98) | 218 (124) | 198 (100) | 195 (99) | 212 (113) | |
| 65 (34) | 257 (149) | 249 (148) | 258 (152) | 249 (145) | 247 (145) | 268 (157) | |
| 82 (43) | 319 (185) | 312 (186) | 337 (203) | 304 (186) | 297 (185) | 324 (205) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, S.; Sha, Q.; Zhang, S. Gene-Based Association Tests Using New Polygenic Risk Scores and Incorporating Gene Expression Data. Genes 2022, 13, 1120. https://doi.org/10.3390/genes13071120
Yan S, Sha Q, Zhang S. Gene-Based Association Tests Using New Polygenic Risk Scores and Incorporating Gene Expression Data. Genes. 2022; 13(7):1120. https://doi.org/10.3390/genes13071120
Chicago/Turabian StyleYan, Shijia, Qiuying Sha, and Shuanglin Zhang. 2022. "Gene-Based Association Tests Using New Polygenic Risk Scores and Incorporating Gene Expression Data" Genes 13, no. 7: 1120. https://doi.org/10.3390/genes13071120
APA StyleYan, S., Sha, Q., & Zhang, S. (2022). Gene-Based Association Tests Using New Polygenic Risk Scores and Incorporating Gene Expression Data. Genes, 13(7), 1120. https://doi.org/10.3390/genes13071120