
Integrated sRNA-seq and RNA-seq Analyses Reveal a microRNA Regulation Network Involved in Cold Response in Pisum sativum L.

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Small RNA Sequencing
2.2. sRNA-seq Data Pre-Processing
2.3. Identification of miRNAs and Their Targets
2.4. RNA-seq Data Processing
2.5. Differential miRNA and Gene Expression Analysis
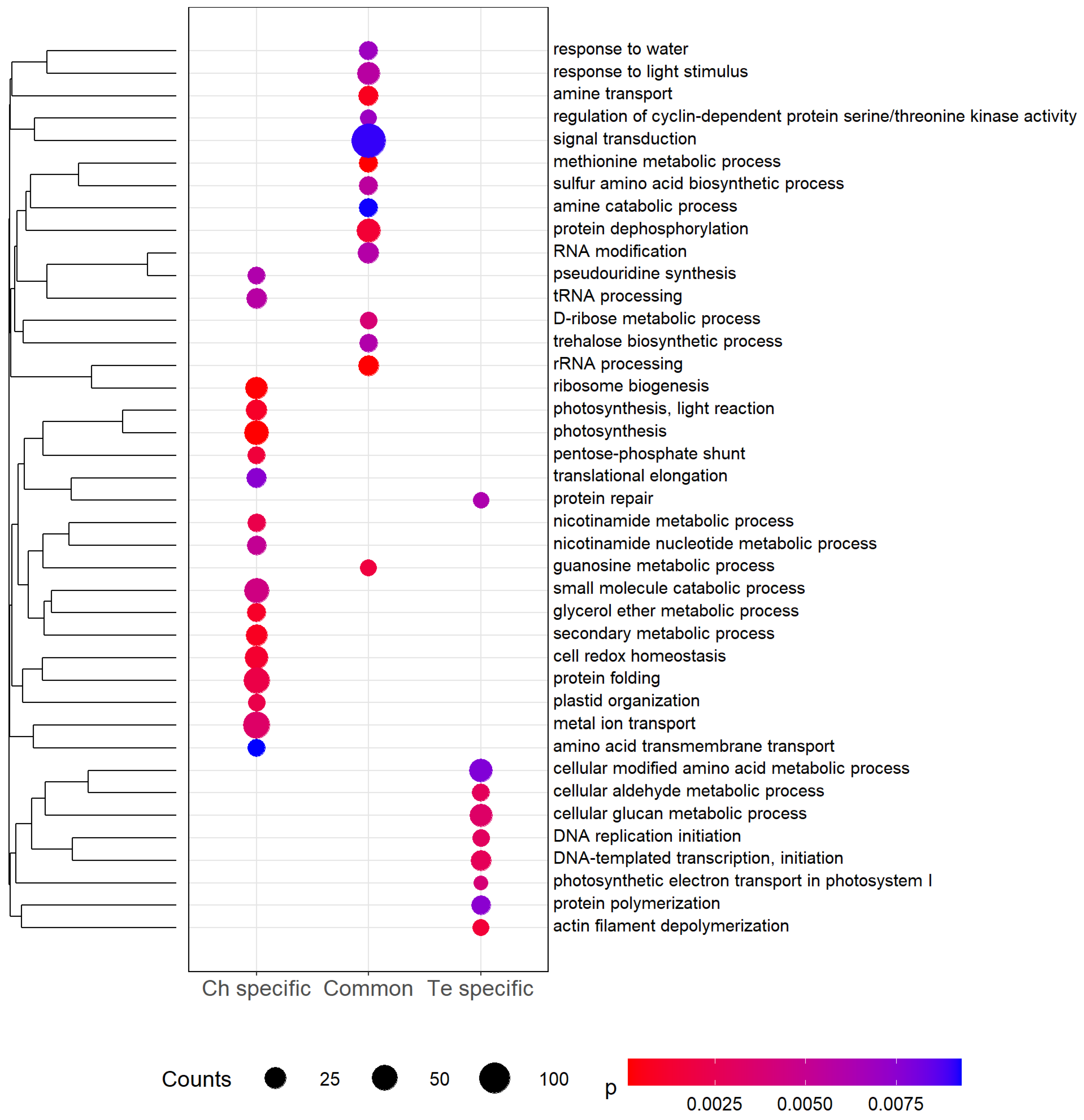
2.6. Gene Ontology Enrichment Analysis
3. Results
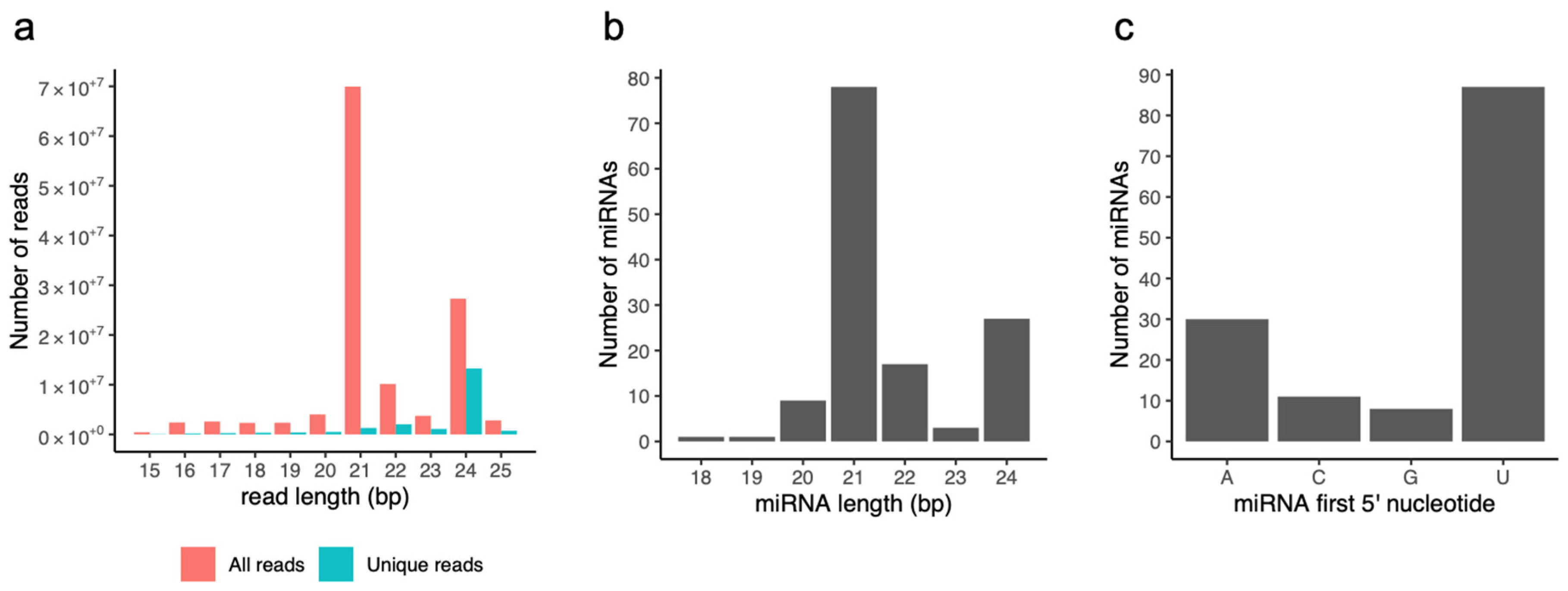
3.1. Small RNA Analysis
3.2. Identification of Conserved and Non-Conserved miRNAs
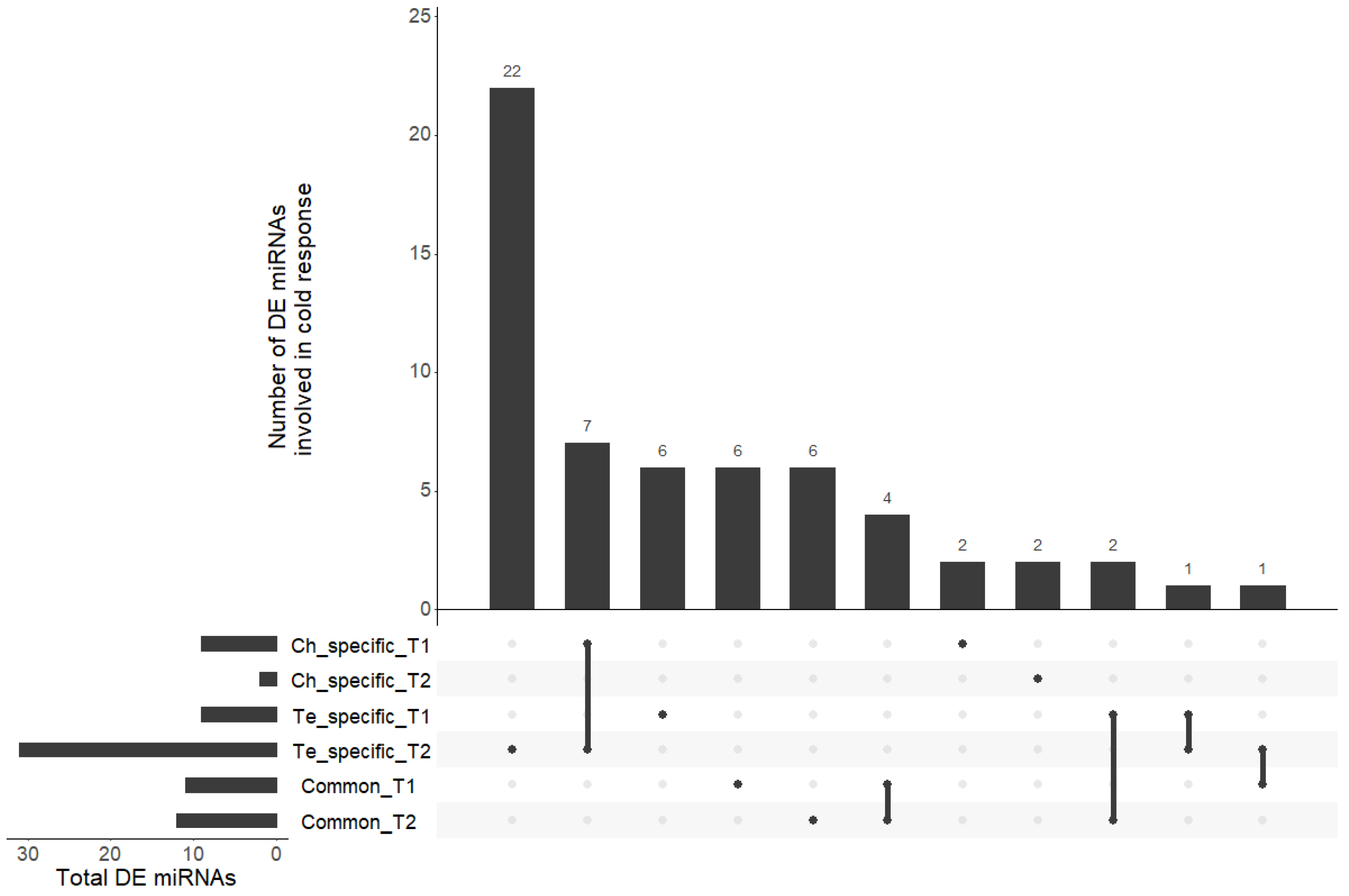
3.3. miRNAs Involved in Cold Response
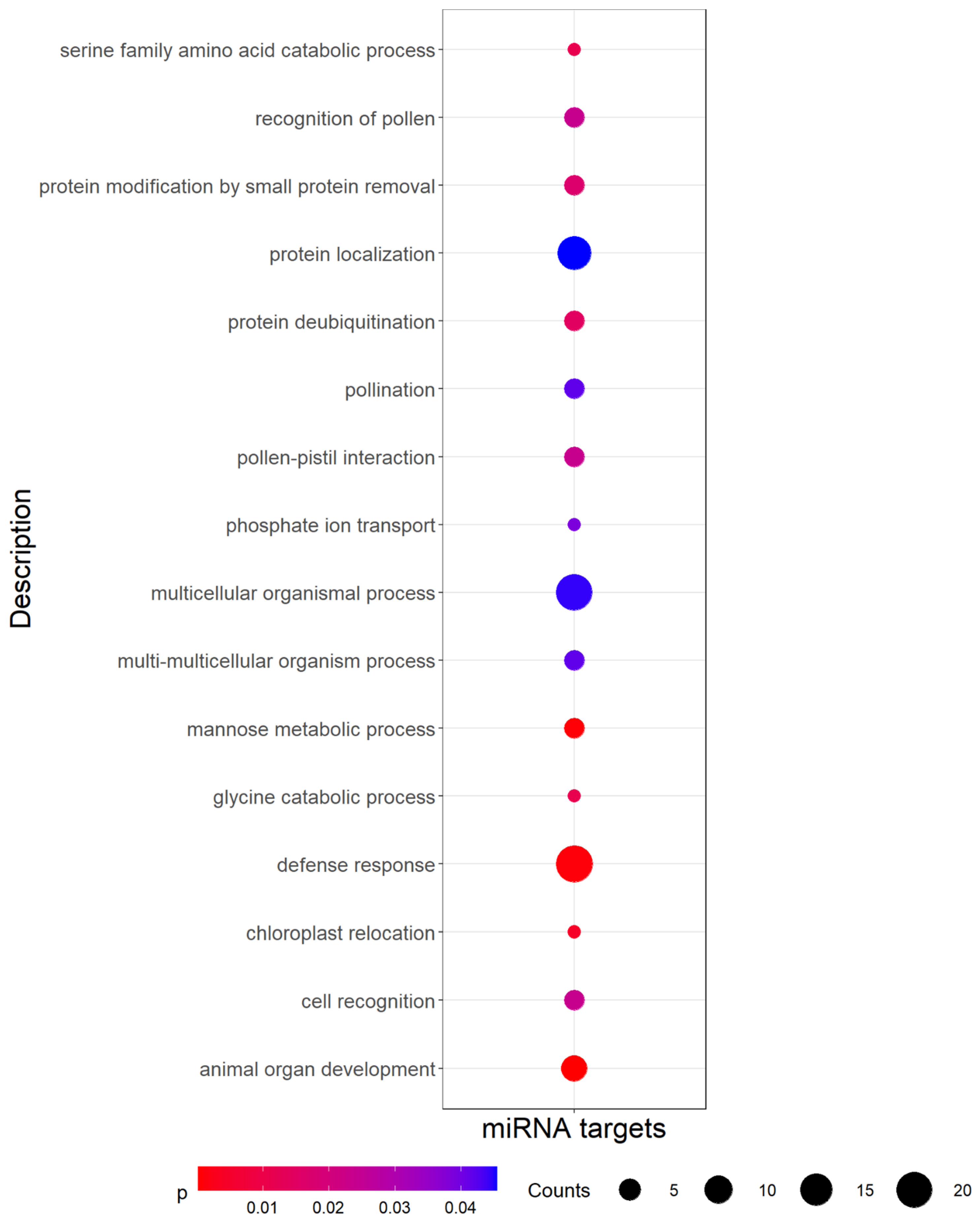
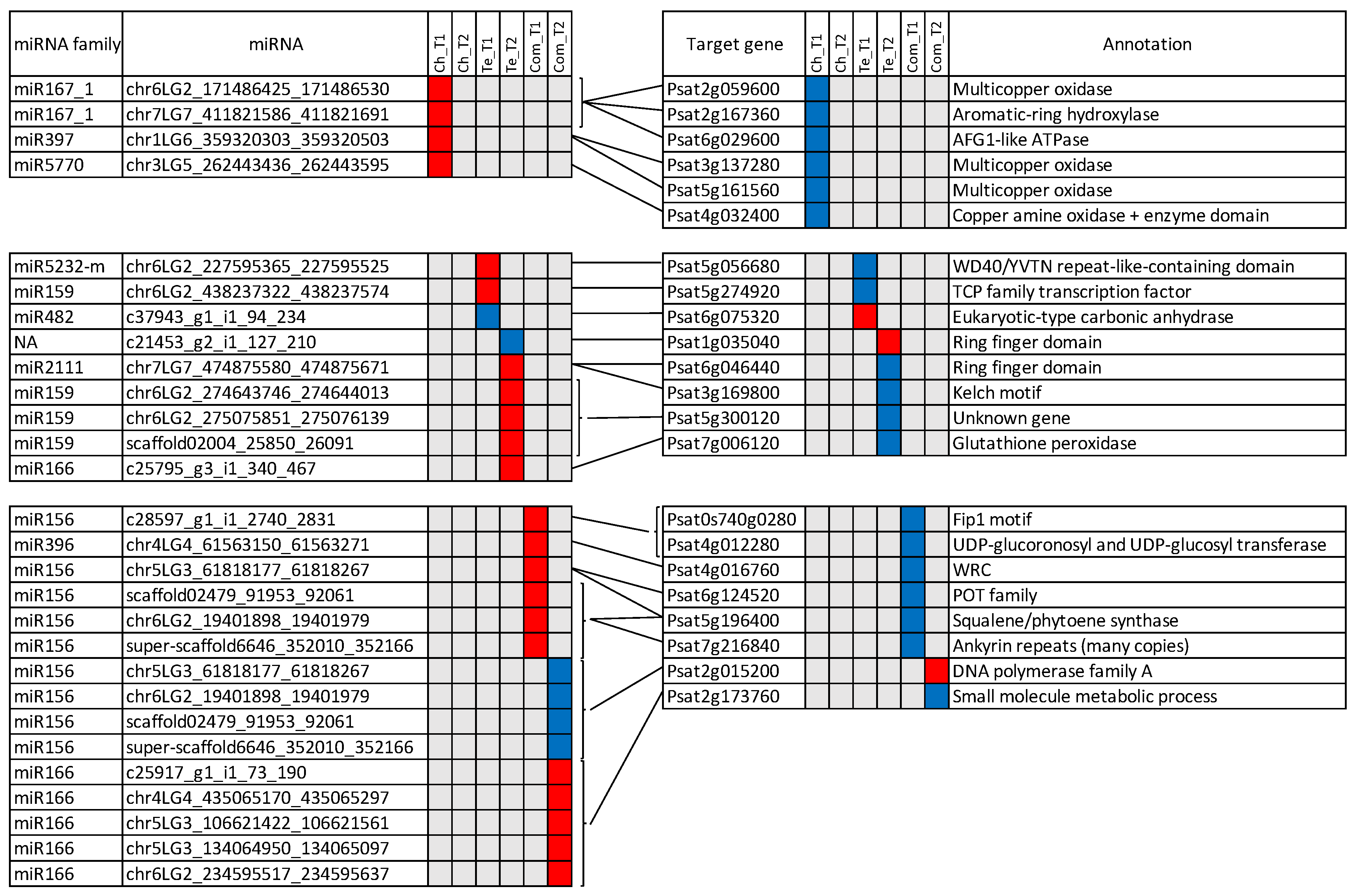
3.4. Identification of miRNA Targets
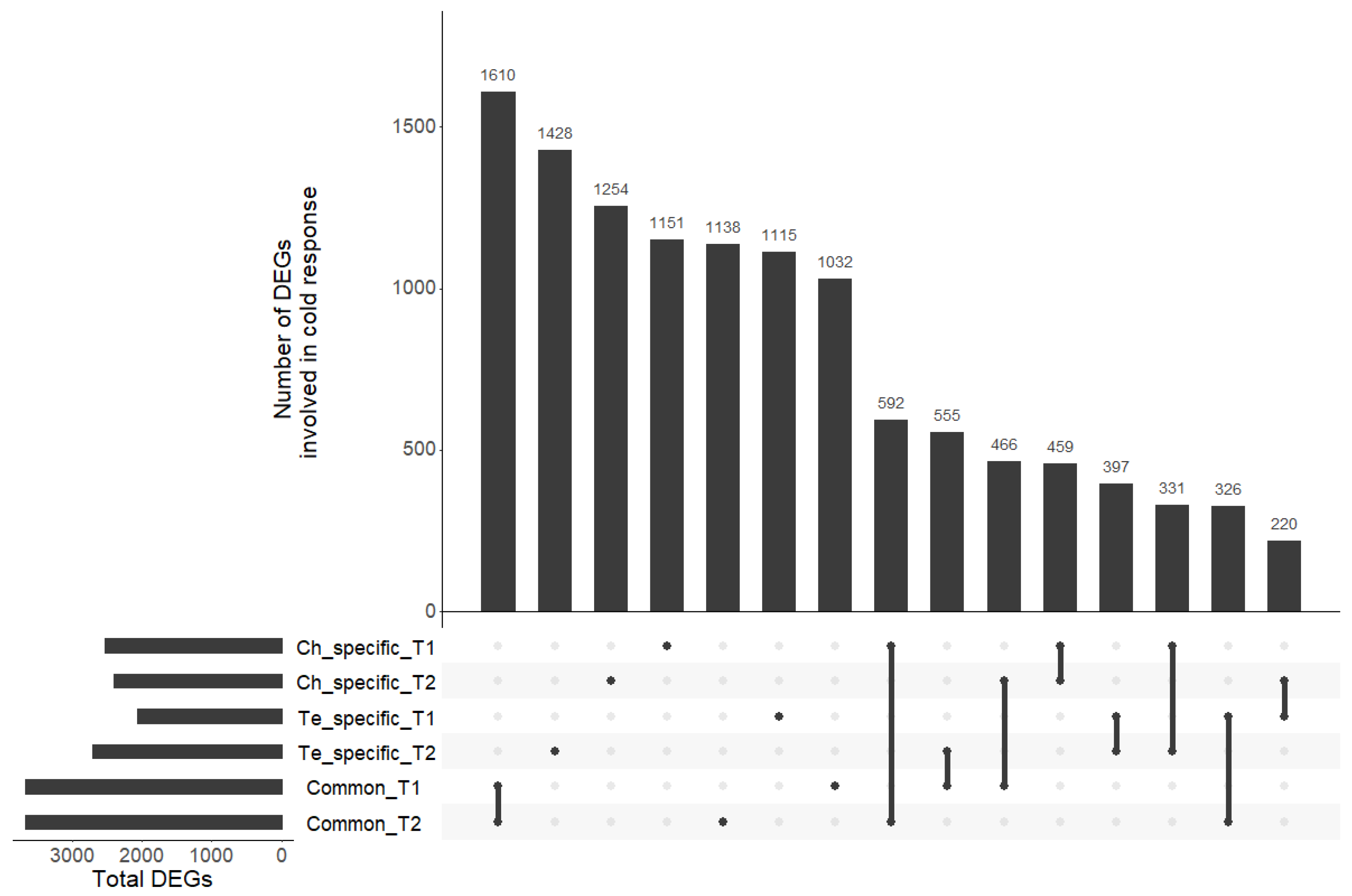
3.5. Differentially Expressed Genes in Response to Cold
3.6. miRNAs and Target mRNAs with Discordant Expression Profiles
4. Discussion
4.1. Metabolic Responses to Cold in Pea
4.2. Regulation of the Cold Response by miRNAs in Pea
4.2.1. Antioxidative Processes Are Predominantly Highlighted in Both Genotypes
4.2.2. Three Conserved miRNA Families Are Highly Represented in the Pea Response to Cold and Could Regulate the Response to Multiple Stresses as Well as Plant Morphology and Phenology
4.2.3. Laccases Are Specifically Involved in Cold Adaptation of the Tolerant Genotype Ch
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferreira, H.; Vasconcelos, M.; Gil, A.M.; Pinto, E. Benefits of pulse consumption on metabolism and health: A systematic review of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2021, 61, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Vaz Patto, M.C.; Amarowicz, R.; Aryee, A.N.A.; Boye, J.I.; Chung, H.-J.; Martín-Cabrejas, M.A.; Domoney, C. Achievements and Challenges in Improving the Nutritional Quality of Food Legumes. Crit. Rev. Plant Sci. 2015, 34, 105–143. [Google Scholar] [CrossRef]
- Jeuffroy, M.H.; Baranger, E.; Carrouee, B.; de Chezelles, E.; Gosme, M.; Henault, C.; Schneider, A.; Cellier, P. Nitrous oxide emissions from crop rotations including wheat, oilseed rape and dry peas. Biogeosciences 2013, 10, 1787–1797. [Google Scholar] [CrossRef] [Green Version]
- Stagnari, F.; Maggio, A.; Galieni, A.; Pisante, M. Multiple benefits of legumes for agriculture sustainability: An overview. Chem. Biol. Technol. Agric. 2017, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Voisin, A.S.; Gueguen, J.; Huyghe, C.; Jeuffroy, M.H.; Magrini, M.B.; Meynard, J.M.; Mougel, C.; Pellerin, S.; Pelzer, E. Legumes for feed, food, biomaterials and bioenergy in Europe: A review. Agron. Sustain. Dev. 2014, 34, 361–380. [Google Scholar] [CrossRef]
- Duc, G.; Agrama, H.; Bao, S.Y.; Berger, J.; Bourion, V.; De Ron, A.M.; Gowda, C.L.L.; Mikic, A.; Millot, D.; Singh, K.B.; et al. Breeding Annual Grain Legumes for Sustainable Agriculture: New Methods to Approach Complex Traits and Target New Cultivar Ideotypes. Crit. Rev. Plant Sci. 2015, 34, 381–411. [Google Scholar] [CrossRef] [Green Version]
- Lejeune-Hénaut, I.; Hanocq, E.; Béthencourt, L.; Fontaine, V.; Delbreil, B.; Morin, J.; Petit, A.; Devaux, R.; Boilleau, M.; Stempniak, J.J.; et al. The flowering locus Hr colocalizes with a major QTL affecting winter frost tolerance in Pisum sativum L. Theor. Appl. Genet. 2008, 116, 1105–1116. [Google Scholar] [CrossRef]
- Beji, S.; Fontaine, V.; Devaux, R.; Thomas, M.; Negro, S.S.; Bahrman, N.; Siol, M.; Aubert, G.; Burstin, J.; Hilbert, J.-L.; et al. Genome-wide association study identifies favorable SNP alleles and candidate genes for frost tolerance in pea. BMC Genom. 2020, 21, 536. [Google Scholar] [CrossRef]
- Grimaud, F.; Renaut, J.; Dumont, E.; Sergeant, K.; Lucau-Danila, A.; Blervacq, A.-S.; Sellier, H.; Bahrman, N.; Lejeune-Hénaut, I.; Delbreil, B.; et al. Exploring chloroplastic changes related to chilling and freezing tolerance during cold acclimation of pea (Pisum sativum L.). J. Proteom. 2013, 80, 145–159. [Google Scholar] [CrossRef]
- Legrand, S.; Marque, G.; Blassiau, C.; Bluteau, A.; Canoy, A.-S.; Fontaine, V.; Jaminon, O.; Bahrman, N.; Mautord, J.; Morin, J.; et al. Combining gene expression and genetic analyses to identify candidate genes involved in cold responses in pea. J. Plant Physiol. 2013, 170, 1148–1157. [Google Scholar] [CrossRef]
- Bahrman, N.; Hascoët, E.; Jaminon, O.; Dépta, F.; Hû, J.-F.; Bouchez, O.; Lejeune-Hénaut, I.; Delbreil, B.; Legrand, S. Identification of Genes Differentially Expressed in Response to Cold in Pisum sativum Using RNA Sequencing Analyses. Plants 2019, 8, 288. [Google Scholar] [CrossRef] [Green Version]
- Rogers, K.; Chen, X.M. Biogenesis, Turnover, and Mode of Action of Plant MicroRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.R.; Khare, T.; Kumar, V. Recent trends and advances in identification and functional characterization of plant miRNAs. Acta Physiol. Plant. 2020, 42, 25. [Google Scholar] [CrossRef]
- Chaudhary, S.; Grover, A.; Sharma, P.C. MicroRNAs: Potential Targets for Developing Stress-Tolerant Crops. Life 2021, 11, 289. [Google Scholar] [CrossRef] [PubMed]
- Bhogireddy, S.; Mangrauthia, S.K.; Kumar, R.; Pandey, A.K.; Singh, S.; Jain, A.; Budak, H.; Varshney, R.K.; Kudapa, H. Regulatory non-coding RNAs: A new frontier in regulation of plant biology. Funct. Integr. Genom. 2021, 21, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Rossi, R.; Paesano, L.; Marmiroli, N.; Marmiroli, M. miRNA regulation and stress adaptation in plants. Environ. Exp. Bot. 2021, 184, 13. [Google Scholar] [CrossRef]
- Liu, Q.; Yan, S.J.; Yang, T.F.; Zhang, S.H.; Chen, Y.Q.; Liu, B. Small RNAs in regulating temperature stress response in plants. J. Integr. Plant Biol. 2017, 59, 774–791. [Google Scholar] [CrossRef] [Green Version]
- Megha, S.; Basu, U.; Kav, N.N.V. Regulation of low temperature stress in plants by microRNAs. Plant Cell Environ. 2018, 41, 1–15. [Google Scholar] [CrossRef]
- Dong, C.H.; Pei, H.X. Over-expression of miR397 improves plant tolerance to cold stress in Arabidopsis thaliana. J. Plant Biol. 2014, 57, 209–217. [Google Scholar] [CrossRef]
- Chen, L.; Luan, Y.S.; Zhai, J.M. Sp-miR396a-5p acts as a stress-responsive genes regulator by conferring tolerance to abiotic stresses and susceptibility to Phytophthora nicotianae infection in transgenic tobacco. Plant Cell Rep. 2015, 34, 2013–2025. [Google Scholar] [CrossRef]
- Zhang, X.N.; Wang, W.; Wang, M.; Zhang, H.Y.; Liu, J.H. The miR396b of Poncirus trifoliata Functions in Cold Tolerance by Regulating ACC Oxidase Gene Expression and Modulating Ethylene-Polyamine Homeostasis. Plant Cell Physiol. 2016, 57, 1865–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreplak, J.; Madoui, M.A.; Capal, P.; Novak, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- BioProject. PRJNA543764. Available online: https://www.ncbi.nlm.nih.gov/bioproject/543764 (accessed on 11 March 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- FastQC. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 April 2020).
- Langmead, B. Aligning Short Sequencing Reads with Bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.17.11–11.17.14. [Google Scholar] [CrossRef]
- Guigon, I.; Legrand, S.; Berthelot, J.F.; Bini, S.; Lanselle, D.; Benmounah, M.; Touzet, H. miRkwood: A tool for the reliable identification of microRNAs in plant genomes. BMC Genom. 2019, 20, 532. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Fahlgren, N.; Carrington, J.C. MiRNA Target Prediction in Plants. Methods Mol. Biol. Clifton NJ 2010, 592, 51–57. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Pysam. Available online: https://github.com/pysam-developers/pysam (accessed on 22 April 2020).
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexa, A.; Rahnenfuhrer, J. TopGO: Enrichment Analysis for Gene Ontology, R Package version 2.42.0; Bioconductor 2020. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 11 March 2022).
- Zhao, C.G.; Wang, Z. GOGO: An improved algorithm to measure the semantic similarity between gene ontology terms. Sci. Rep. 2018, 8, 15107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, I.R.; Zhang, X.Y.; Lu, C.; Johnson, L.; Meyers, B.C.; Green, P.J.; Jacobsen, S.E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006, 38, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010, 61, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, T.Z.; Zhao, M.G.; Tian, Q.Y.; Zhang, W.H. Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 2012, 235, 375–386. [Google Scholar] [CrossRef]
- Liu, Y.X.; Wang, M.; Wang, X.J. Endogenous small RNA clusters in plants. Genom. Proteom. Bioinform. 2014, 12, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.X.; Wang, Y.Y.; Gao, X.K.; Liu, C.Y.; Gai, S.P. Identification and characterization of microRNAs in tree peony during chilling induced dormancy release by high-throughput sequencing. Sci. Rep. 2018, 8, 4537. [Google Scholar] [CrossRef] [Green Version]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and Functional Diversification of MIRNA Genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, J.; Bryant, G. Freezing, drying, and/or vitrification of membrane-solute-water systems. Cryobiology 1999, 39, 103–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iordachescu, M.; Imai, R. Trehalose biosynthesis in response to abiotic stresses. J. Integr. Plant Biol. 2008, 50, 1223–1229. [Google Scholar] [CrossRef]
- Kreps, J.A.; Wu, Y.J.; Chang, H.S.; Zhu, T.; Wang, X.; Harper, J.F. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002, 130, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Zagorchev, L.; Seal, C.E.; Kranner, I.; Odjakova, M. A Central Role for Thiols in Plant Tolerance to Abiotic Stress. Int. J. Mol. Sci. 2013, 14, 7405–7432. [Google Scholar] [CrossRef] [Green Version]
- Amini, S.; Maali-Amiri, R.; Kazemi-Shahandashti, S.S.; Lopez-Gomez, M.; Sadeghzadeh, B.; Sobhani-Najafabadi, A.; Kariman, K. Effect of cold stress on polyamine metabolism and antioxidant responses in chickpea. J. Plant Physiol. 2021, 258–259, 153387. [Google Scholar] [CrossRef]
- Bashir, K.; Ahmad, Z.; Kobayashi, T.; Seki, M.; Nishizawa, N.K. Roles of subcellular metal homeostasis in crop improvement. J. Exp. Bot. 2021, 72, 2083–2098. [Google Scholar] [CrossRef]
- Boston, R.S.; Viitanen, P.V.; Vierling, E. Molecular chaperones and protein folding in plants. Plant Mol. Biol. 1996, 32, 191–222. [Google Scholar] [CrossRef]
- Sung, D.Y.; Vierling, E.; Guy, C.L. Comprehensive expression profile analysis of the Arabidopsis hsp70 gene family. Plant Physiol. 2001, 126, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.P.; Pan, F.L.; Lou, X.P.; Wang, D.B.; Yang, C.; Zhang, B.Q.; Zhang, H.Y. Genome-wide identification and characterization of Hsp70 gene family in Nicotiana tabacum. Mol. Biol. Rep. 2019, 46, 1941–1954. [Google Scholar] [CrossRef] [PubMed]
- Ambroise, V.; Legay, S.; Guerriero, G.; Hausman, J.-F.; Cuypers, A.; Sergeant, K. The Roots of Plant Frost Hardiness and Tolerance. Plant Cell Physiol. 2019, 61, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvar, B.L.; Sangwan, V.; Omann, F.; Dhindsa, R.S. Early steps in cold sensing by plant cells: The role of actin cytoskeleton and membrane fluidity. Plant J. 2000, 23, 785–794. [Google Scholar] [CrossRef]
- Sangwan, V.; Foulds, I.; Singh, J.; Dhindsa, R.S. Cold-activation of Brassica napus BN115 promoter is mediated by structural changes in membranes and cytoskeleton, and requires Ca2+ influx. Plant J. 2001, 27, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokorna, J.; Schwarzerova, K.; Zelenkova, S.; Petrasek, J.; Janotova, I.; Capkova, V.; Opatrny, Z. Sites of actin filament initiation and reorganization in cold-treated tobacco cells. Plant Cell Environ. 2004, 27, 641–653. [Google Scholar] [CrossRef]
- Wang, L.X.; Sadeghnezhad, E.; Riemann, M.; Nick, P. Microtubule dynamics modulate sensing during cold acclimation in grapevine suspension cells. Plant Sci. 2019, 280, 18–30. [Google Scholar] [CrossRef]
- Rao, M.J.; Xu, Y.T.; Huang, Y.; Tang, X.M.; Deng, X.X.; Xu, Q. Ectopic expression of citrus UDP-GLUCOSYL TRANSFERASE gene enhances anthocyanin and proanthocyanidins contents and confers high light tolerance in Arabidopsis. BMC Plant Biol. 2019, 19, 603. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.Z.; Zhang, D.Y.; Zhou, L.; Zhang, X.Z.; Liao, J.R.; Duan, Y.; Wen, B.; Ma, Y.C.; Wang, Y.H.; Fang, W.P.; et al. Transcriptomic and metabolomic profiling of Camellia sinensis L. cv. ‘Suchazao’ exposed to temperature stresses reveals modification in protein synthesis and photosynthetic and anthocyanin biosynthetic pathways. Tree Physiol. 2019, 39, 1583–1599. [Google Scholar] [CrossRef]
- Zhang, Y.C.; He, R.R.; Lian, J.P.; Zhou, Y.F.; Zhang, F.; Li, Q.F.; Yu, Y.; Feng, Y.Z.; Yang, Y.W.; Lei, M.Q.; et al. OsmiR528 regulates rice-pollen intine formation by targeting an uclacyanin to influence flavonoid metabolismz. Proc. Natl. Acad. Sci. USA 2020, 117, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Statham, C.M.; Crowden, R.K.; Harborne, J.B. Biochemical genetics of pigmentation in Pisum sativum. Phytochemistry 1972, 11, 1083–1088. [Google Scholar] [CrossRef]
- Marx, G.A.; Weeden, N.F.; Muehlbauer, F.J. A-2: A new locus controlling anthocyanin production in Pisum. Pisum Newsl. 1989, 21, 35–36. [Google Scholar]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.E.J.; Frew, T.J.; Murray, S.R.; Hofer, J.M.I.; Jacobs, J.M.E.; et al. Identification of Mendel’s White Flower Character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, C.; Ambrose, M.J.; Turner, L.; Hill, L.; Ellis, T.H.N.; Hofer, J.M.I. The b Gene of Pea Encodes a Defective Flavonoid 3′,5′-Hydroxylase, and Confers Pink Flower Color. Plant Physiol. 2012, 159, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demmig-Adams, B.; Adams, W.W. Antioxidants in photosynthesis and human nutrition. Science 2002, 298, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Flowerika; Alok, A.; Kumar, J.; Thakur, N.; Pandey, A.; Pandey, A.K.; Upadhyay, S.K.; Tiwari, S. Characterization and Expression Analysis of Phytoene Synthase from Bread Wheat (Triticum aestivum L.). PLoS ONE 2016, 11, e0162443. [Google Scholar] [CrossRef]
- Li, F.Q.; Vallabhaneni, R.; Yu, J.; Rocheford, T.; Wurtzel, E.T. The maize phytoene synthase gene family: Overlapping roles for carotenogenesis in endosperm, photomorphogenesis, and thermal stress tolerance. Plant Physiol. 2008, 147, 1334–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Dogru, A.; Cakirlar, H. Effects of leaf age on chlorophyll fluorescence and antioxidant enzymes activity in winter rapeseed leaves under cold acclimation conditions. Braz. J. Bot. 2020, 43, 11–20. [Google Scholar] [CrossRef]
- Vo, K.T.X.; Kim, C.Y.; Chandran, A.K.N.; Jung, K.H.; An, G.; Jeon, J.S. Molecular insights into the function of ankyrin proteins in plants. J. Plant Biol. 2015, 58, 271–284. [Google Scholar] [CrossRef]
- Chen, L.; Hellmann, H. Plant E3 Ligases: Flexible Enzymes in a Sessile World. Mol. Plant 2013, 6, 1388–1404. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.M.; Meng, Q.L.; Zhang, H.S.; Huang, J. Knock-down of a RING finger gene confers cold tolerance. Bioengineered 2016, 7, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Xu, Y.; Jiang, J.; Zhang, F.; Ma, L.; Wu, D.; Wang, Y.; Sun, W. Identification of cold stress responsive microRNAs in two winter turnip rape (Brassica rapa L.) by high throughput sequencing. BMC Plant Biol. 2018, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zhang, R.; Zhang, S.; Li, Y.; Gao, J.; Han, X.; Chen, M.; Wang, J.; Li, W.; Li, G. Response of microRNAs to cold treatment in the young spikes of common wheat. BMC Genom. 2017, 18, 212. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.J.; Liu, Y.; Li, W.; Song, L.L.; Zhang, J.; Guo, C.H. Genome-Wide Investigation of MicroRNAs and Their Targets in Response to Freezing Stress in Medicago sativa L., Based on High-Throughput Sequencing. G3 Genes Genom. Genet. 2016, 6, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.G.; Shan, J.X.; Shi, M.; Gao, J.P.; Lin, H.X. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014, 80, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Forbes, K.P.; Addepalli, B.; Hunt, A.G. An Arabidopsis Fip1 homolog interacts with RNA and provides conceptual links with a number of other polyadenylation factor subunits. J. Biol. Chem. 2006, 281, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Tellez-Robledo, B.; Manzano, C.; Saez, A.; Navarro-Neila, S.; Silva-Navas, J.; de Lorenzo, L.; Gonzalez-Garcia, M.P.; Toribio, R.; Hunt, A.G.; Baigorri, R.; et al. The polyadenylation factor FIP1 is important for plant development and root responses to abiotic stresses. Plant J. 2019, 99, 1203–1219. [Google Scholar] [CrossRef]
- Xin, M.M.; Wang, Y.; Yao, Y.Y.; Xie, C.J.; Peng, H.R.; Ni, Z.F.; Sun, Q.X. Diverse set of microRNAs are responsive to powdery mildew infection and heat stress in wheat (Triticum aestivum L.). BMC Plant Biol. 2010, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Boutet, G.; Lavaud, C.; Coyne, C.J.; Philippe, D.; Lejeune-Henaut, I.; Lesné, A.; Pilet-Nayel, M.-L.; Baranger, A. Identification of regions in the pea genome controlling both stress resistance and developmental traits. In Proceedings of the ICLGG 2019, Dijon, France, 13 May 2019. [Google Scholar]
- McCaig, B.C.; Meagher, R.B.; Dean, J.F.D. Gene structure and molecular analysis of the laccase-like multicopper oxidase (LMCO) gene family in Arabidopsis thaliana. Planta 2005, 221, 619–636. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.Z.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Y.; Gong, H.Y.; Cao, L.F.; Hou, Y.J.; Qu, S.C. MicroRNA397b negatively regulates resistance of Malus hupehensis to Botryosphaeria dothidea by modulating MhLAC7 involved in lignin biosynthesis. Plant Sci. 2020, 292, 110390. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| List of DEGs | DEGs at T1 | DEGs at T2 | |
|---|---|---|---|
| Te specific cold responsive genes | (−) 1171 (+) 887 | (−) 1569 (+) 1142 | |
| Ch specific cold responsive genes | (−) 1332 (+) 1201 | (−) 1162 (+) 1237 | |
| Te and Ch common cold responsive genes | Concordant genes | (−) 1719 (+) 1921 | (−) 2000 (+) 1613 |
| Discordant genes | 12 (+) Te & (−) Ch 11 (−) Te & (+) Ch | 19 (+) Te & (−) Ch 34 (−) Te & (+) Ch | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurier, M.; Drouaud, J.; Bahrman, N.; Rau, A.; Lejeune-Hénaut, I.; Delbreil, B.; Legrand, S. Integrated sRNA-seq and RNA-seq Analyses Reveal a microRNA Regulation Network Involved in Cold Response in Pisum sativum L. Genes 2022, 13, 1119. https://doi.org/10.3390/genes13071119
Mazurier M, Drouaud J, Bahrman N, Rau A, Lejeune-Hénaut I, Delbreil B, Legrand S. Integrated sRNA-seq and RNA-seq Analyses Reveal a microRNA Regulation Network Involved in Cold Response in Pisum sativum L. Genes. 2022; 13(7):1119. https://doi.org/10.3390/genes13071119
Chicago/Turabian StyleMazurier, Mélanie, Jan Drouaud, Nasser Bahrman, Andrea Rau, Isabelle Lejeune-Hénaut, Bruno Delbreil, and Sylvain Legrand. 2022. "Integrated sRNA-seq and RNA-seq Analyses Reveal a microRNA Regulation Network Involved in Cold Response in Pisum sativum L." Genes 13, no. 7: 1119. https://doi.org/10.3390/genes13071119
APA StyleMazurier, M., Drouaud, J., Bahrman, N., Rau, A., Lejeune-Hénaut, I., Delbreil, B., & Legrand, S. (2022). Integrated sRNA-seq and RNA-seq Analyses Reveal a microRNA Regulation Network Involved in Cold Response in Pisum sativum L. Genes, 13(7), 1119. https://doi.org/10.3390/genes13071119