
Metagenomics Reveals Specific Microbial Features in Males with Semen Alterations

 ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patients’ Enrollment and Sample Collection
2.2. DNA Extraction and 16S rRNA Analysis
2.3. Bioinformatic Analysis
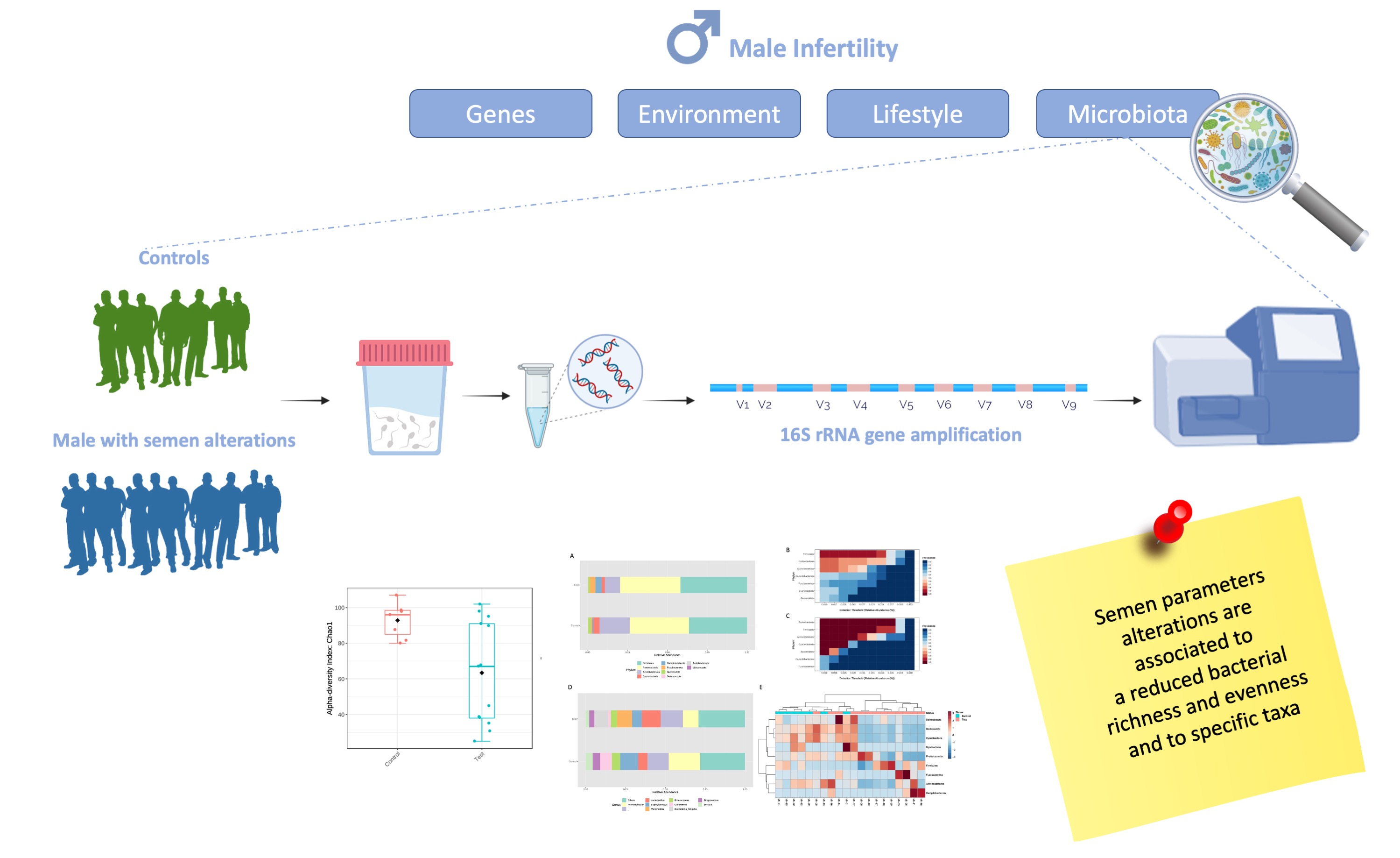
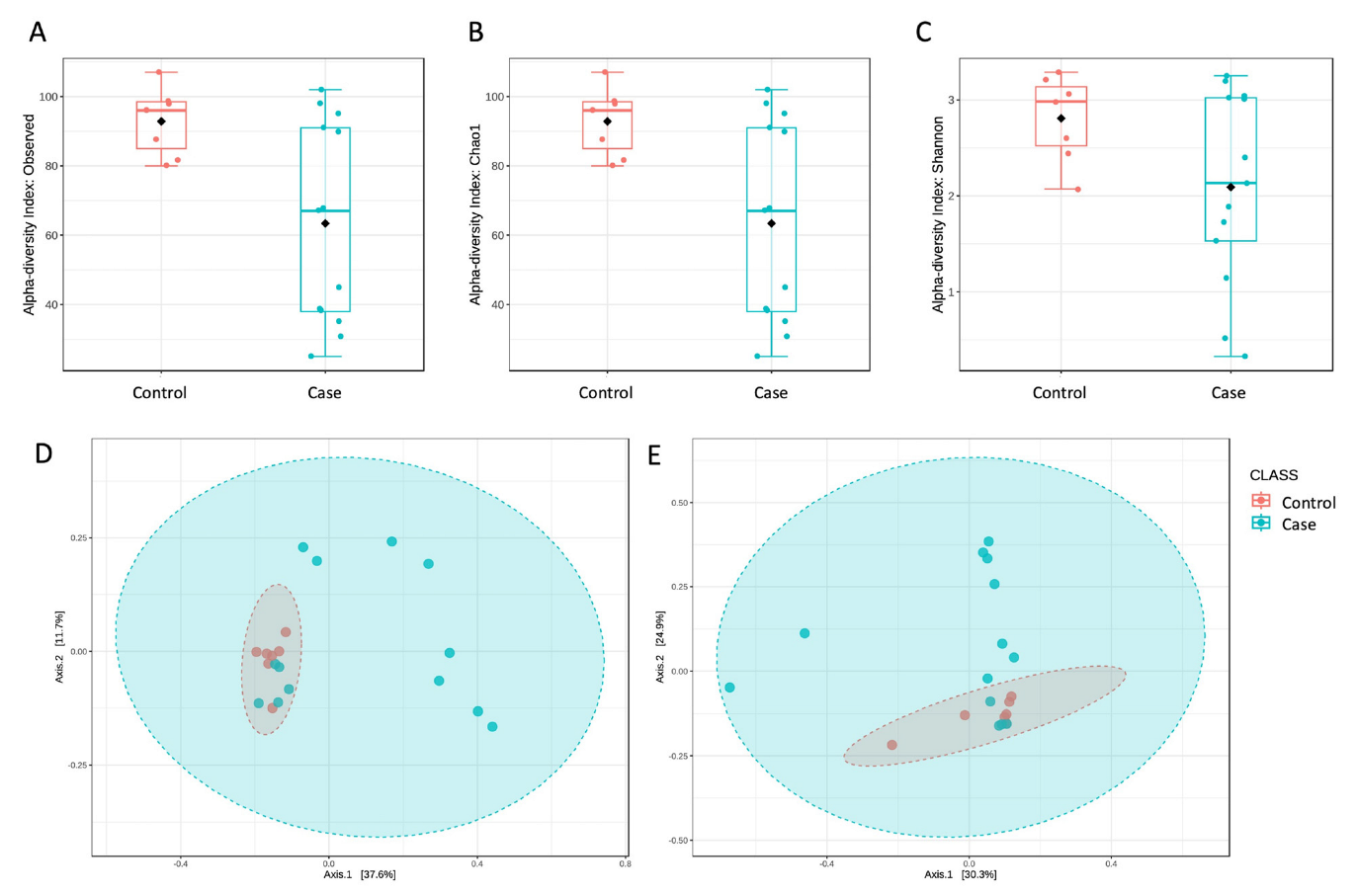
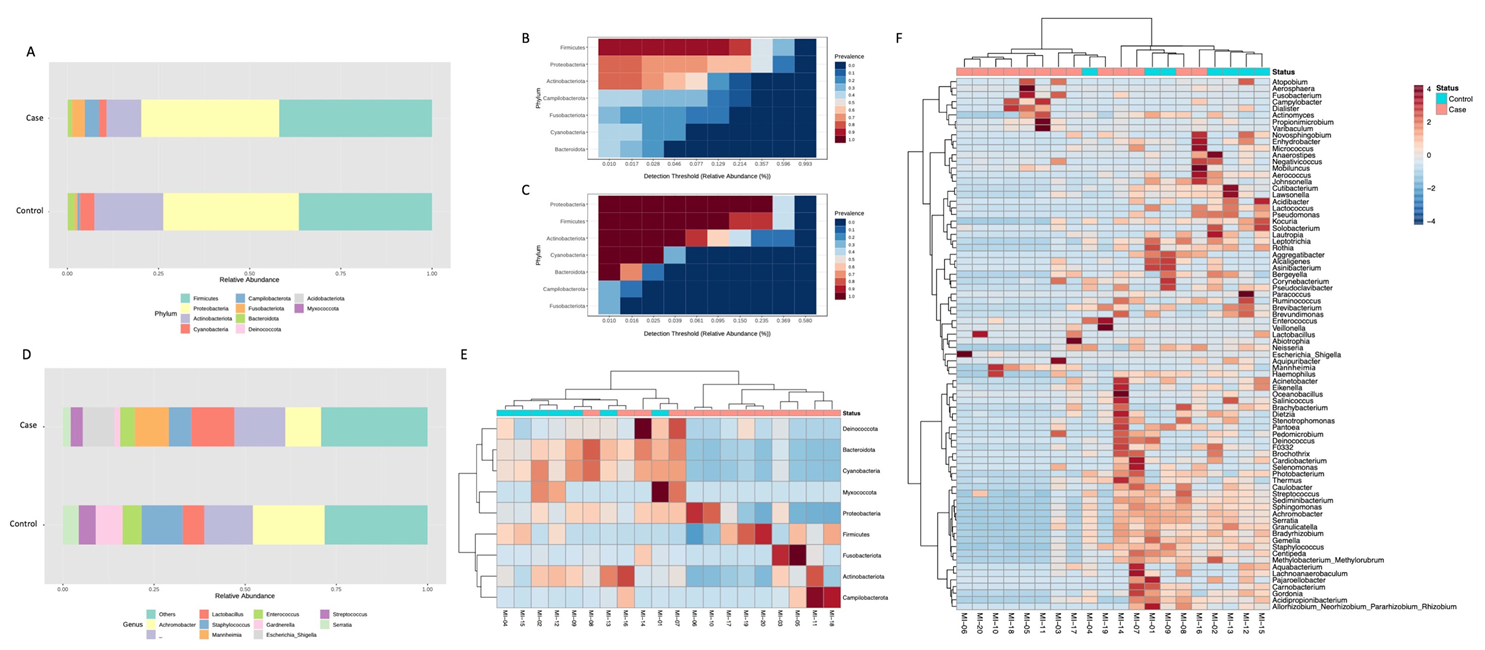
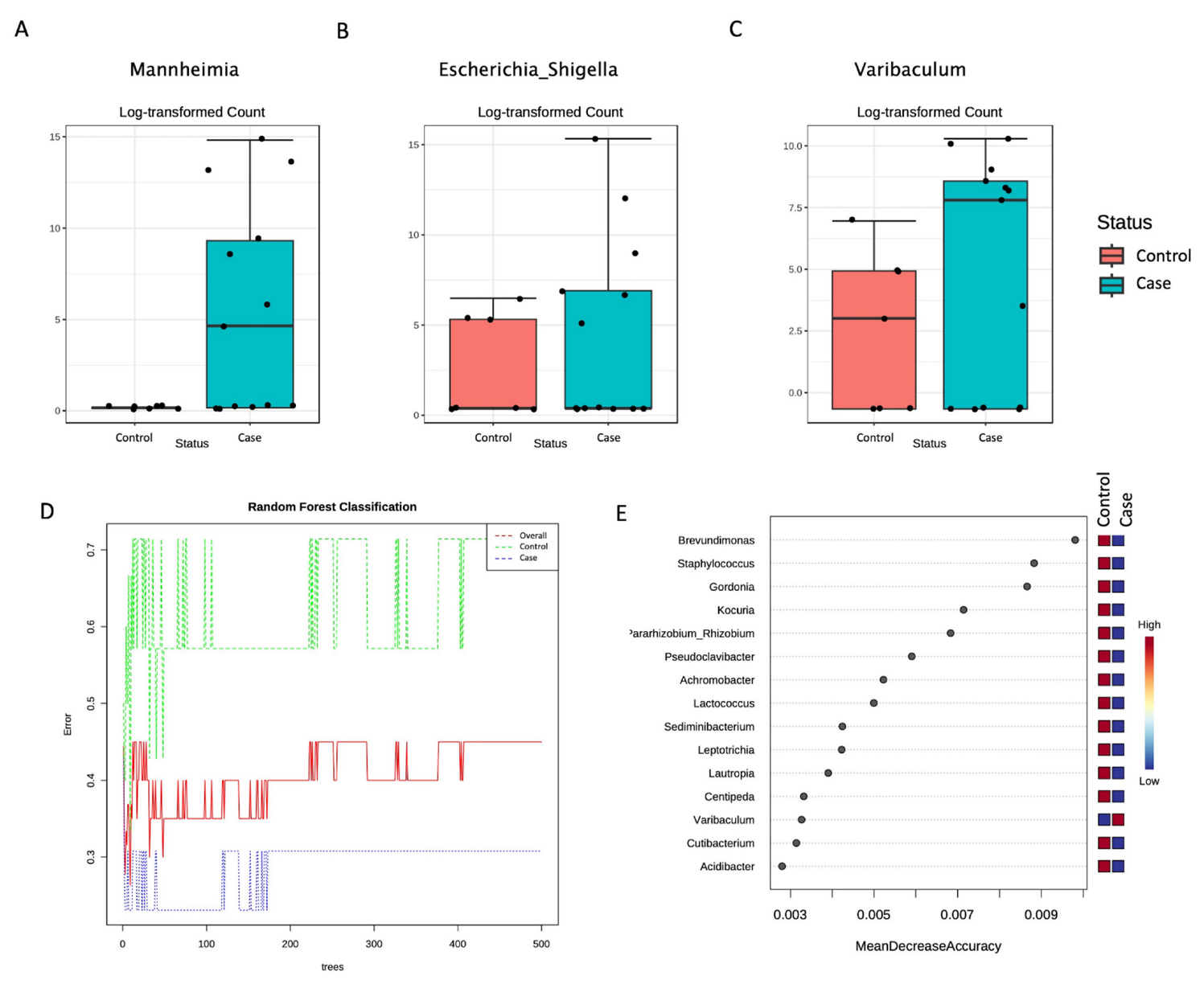
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Agarwal, A.; Parekh, N.; Panner Selvam, M.K.; Henkel, R.; Shah, R.; Homa, S.T.; Ramasamy, R.; Ko, E.; Tremellen, K.; Esteves, S.; et al. Male Oxidative Stress Infertility (MOSI): Proposed Terminology and Clinical Practice Guidelines for Management of Idiopathic Male Infertility. World J. Men’s Health 2019, 37, 296–312. [Google Scholar] [CrossRef]
- Krausz, C. Male infertility: Pathogenesis and clinical diagnosis. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, R.; Veneruso, I.; Cariati, F.; D’Argenio, V. Microbiota and Human Reproduction: The Case of Male Infertility. High-Throughput 2020, 9, 10. [Google Scholar] [CrossRef]
- D’Argenio, V.; Cariati, F.; Tomaiuolo, R. One4Two®: An Integrated Molecular Approach to Optimize Infertile Couples’ Journey. Genes 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- D′Argenio, V.; Dittfeld, L.; Lazzeri, P.; Tomaiuolo, R.; Tasciotti, E. Unraveling the Balance between Genes, Microbes, Lifestyle and the Environment to Improve Healthy Reproduction. Genes 2021, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Cariati, F.; D’Argenio, V.; Tomaiuolo, R. The evolving role of genetic tests in reproductive medicine. J. Transl. Med. 2019, 17, 1–33. [Google Scholar] [CrossRef] [Green Version]
- WHO. WHO Laboratory Manual for the Examination and Processing of Human Semen, 6th ed.; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Tomaiuolo, G.; Fellico, F.; Preziosi, V.; Guido, S. Semen rheology and its relation to male infertility. Interface Focus 2022, 12, 20220048. [Google Scholar] [CrossRef]
- Pagliuca, C.; Cariati, F.; Bagnulo, F.; Scaglione, E.; Carotenuto, C.; Farina, F.; D’Argenio, V.; Carraturo, F.; D’aprile, P.; Vitiello, M.; et al. Microbiological Evaluation and Sperm DNA Fragmentation in Semen Samples of Patients Undergoing Fertility Investigation. Genes 2021, 12, 654. [Google Scholar] [CrossRef]
- Tomaiuolo, R.; Veneruso, I.; Cariati, F.; D’Argenio, V. Microbiota and Human Reproduction: The Case of Female Infertility. High-Throughput 2020, 9, 12. [Google Scholar] [CrossRef]
- D’Argenio, V.; Veneruso, I.; Gong, C.; Cecarini, V.; Bonfili, L.; Eleuteri, A.M. Gut Microbiome and Mycobiome Alterations in an In Vivo Model of Alzheimer’s Disease. Genes 2022, 13, 1564. [Google Scholar] [CrossRef]
- Cecarini, V.; Gogoi, O.; Bonfili, L.; Veneruso, I.; Pacinelli, G.; De Carlo, S.; Benvenuti, F.; D’Argenio, V.; Angeletti, M.; Cannella, N.; et al. Modulation of Gut Microbiota and Neuroprotective Effect of a Yeast-Enriched Beer. Nutrients 2022, 14, 2380. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef] [PubMed]
- D’Argenio, V. Human Microbiome Acquisition and Bioinformatic Challenges in Metagenomic Studies. Int. J. Mol. Sci. 2018, 19, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Luo, T.; Chen, T.; Wang, G. Seminal bacterial composition in patients with obstructive and non-obstructive azoo-spermia. Exp. Ther. Med. 2018, 15, 2884–2890. [Google Scholar]
- Weng, S.-L.; Chiu, C.-M.; Lin, F.-M.; Huang, W.-C.; Liang, C.; Yang, T.; Yang, T.-L.; Liu, C.-Y.; Wu, W.-Y.; Chang, Y.-A.; et al. Bacterial Communities in Semen from Men of Infertile Couples: Metagenomic Sequencing Reveals Relationships of Seminal Microbiota to Semen Quality. PLoS ONE 2014, 9, e110152. [Google Scholar] [CrossRef] [Green Version]
- Ibadin, O.K.; Ibeh, I.N. Bacteriospermia and sperm quality in infertile male patient at University of Benin Teaching Hospital, Benin City, Nigeria. Malays. J. Microbiol. 2008, 4, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Sanocka-Maciejewska, D.; Ciupińska, M.; Kurpisz, M. Bacterial infection and semen quality. J. Reprod. Immunol. 2005, 67, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.; Sánchez, R.; Soto, L.; Risopatrón, J.; Villegas, J. Effect of Escherichia coli and its soluble factors on mitochondrial membrane potential, phosphatidylserine translocation, viability, and motility of human spermatozoa. Fertil. Steril. 2010, 94, 619–623. [Google Scholar] [CrossRef]
- Berktas, M.; Aydin, S.; Yilmaz, Y.; Cecen, K.; Bozkurt, H. Sperm motility changes after coincubation with various uropathogenic microorganisms: An in vitro experimental study. Int. Urol. Nephrol. 2008, 40, 383–389. [Google Scholar] [CrossRef]
- De Francesco, M.A.; Negrini, R.; Ravizzola, G.; Galli, P.; Manca, N. Bacterial species present in the lower male genital tract: A five-year retrospective study. Eur. J. Contracept. Reprod. Health Care 2011, 16, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Jarvi, K.; Lacroix, J.-M.; Jain, A.; Dumitru, I.; Heritz, D.; Mittelman, M.W. Polymerase chain reaction-based detection of bacteria in semen. Fertil. Steril. 1996, 66, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, A.A.; Desmarais, B.M.; Yin, H.-Z.; Loverde, J.; Eyre, R.C. Detection and identification of bacterial DNA in semen. Fertil. Steril. 2008, 90, 1744–1756. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Valenzuela, C.; Montes-García, J.F.; Vazquez-Cruz, C.; Zenteno, E.; Pereyra, M.A.; Negrete-Abascal, E. Mannheimia haemolytica OmpH binds fibrinogen and fibronectin and participates in biofilm formation. Microb. Pathog. 2022, 172, 105788. [Google Scholar] [CrossRef]
- Azhar, N.A.; Paul, B.T.; Jesse, F.F.A.; Chung, E.L.T.; Kamarulrizal, M.I.; Mohd Lila, M.A. Seminal and histopathological al-terations in bucks challenged with Mannheimia haemolytica serotype a2 and its LPS endotoxin. Trop. Anim. Health Prod. 2022, 54, 265. [Google Scholar] [CrossRef]
- Corbel, M.J. 35—Yersinia, Yersinia, Pasteurella and Francisella: Plague; Pseudotuberculosis; Mesenteric Adenitis; Pasteurellosis; Tularae-mia. In Medical Microbiology, 18th ed.; Churchill Livingstone: London, UK, 2012; pp. 350–358. [Google Scholar]
- Lau, J.S.Y.; Omaleki, L.; Turni, C.; Barber, S.R.; Browning, G.F.; Francis, M.J.; Graham, M.; Korman, T.M. Human Wound Infection with Mannheimia glucosida following Lamb Bite. J. Clin. Microbiol. 2015, 53, 3374–3376. [Google Scholar] [CrossRef] [Green Version]
- Roier, S.; Fenninger, J.C.; Leitner, D.R.; Rechberger, G.N.; Reidl, J.; Schild, S. Immunogenicity of Pasteurella multocida and Mannheimia haemolytica outer membrane vesicles. Int. J. Med. Microbiol. 2013, 303, 247–256. [Google Scholar] [CrossRef]
- Mändar, R.; Punab, M.; Korrovits, P.; Türk, S.; Ausmees, K.; Lapp, E.; Preem, J.-K.; Oopkaup, K.; Salumets, A.; Truu, J. Seminal microbiome in men with and without prostatitis. Int. J. Urol. 2017, 24, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Hrbáček, J.; Tláskal, V.; Čermák, P.; Hanáček, V.; Zachoval, R. Bladder cancer is associated with decreased urinary microbiota diversity and alterations in microbial community composition. Urol. Oncol. 2023, 41, 107.e15–107.e22. [Google Scholar] [CrossRef]
- Hurst, R.; Meader, E.; Gihawi, A.; Rallapalli, G.; Clark, J.; Kay, G.L.; Webb, M.; Manley, K.; Curley, H.; Walker, H.; et al. Microbiomes of Urine and the Prostate Are Linked to Human Prostate Cancer Risk Groups. Eur. Urol. Oncol. 2022, 5, 412–419. [Google Scholar] [CrossRef]
- Mändar, R.; Punab, M.; Borovkova, N.; Lapp, E.; Kiiker, R.; Korrovits, P.; Metspalu, A.; Krjutškov, K.; Nõlvak, H.; Preem, J.-K.; et al. Complementary seminovaginal microbiome in couples. Res. Microbiol. 2015, 166, 440–447. [Google Scholar] [CrossRef]
- Du Plessis, S.S.; Gokul, S.; Agarwal, A. Semen hyperviscosity: Causes, consequences, and cures. Front Biosci. 2013, 5, 224–231. [Google Scholar]
- Monteiro, C.; Marques, P.I.; Cavadas, B.; Damião, I.; Almeida, V.; Barros, N.; Barros, A.; Carvalho, F.; Gomes, S.; Seixas, S. Characterization of microbiota in male infertility cases uncovers differences in seminal hyperviscosity and oligoasthenotera-tozoospermia possibly correlated with increased prevalence of infectious bacteria. Am. J. Reprod. Immunol. 2018, 79, e12838. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Segura, S.; del Rey, J.; Closa, L.; Garcia-Martínez, I.; Hobeich, C.; Castel, A.B.; Vidal, F.; Benet, J.; Ribas-Maynou, J.; Oliver-Bonet, M. Seminal Microbiota of Idiopathic Infertile Patients and Its Relationship With Sperm DNA Integrity. Front. Cell Dev. Biol. 2022, 10, 937157. [Google Scholar] [CrossRef] [PubMed]
- Baud, D.; Pattaroni, C.; Vulliemoz, N.; Castella, V.; Marsland, B.J.; Stojanov, M. Sperm Microbiota and Its Impact on Semen Parameters. Front. Microbiol. 2019, 10, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhang, J.; Xue, Z.; Zhao, C.; Lei, L.; Wen, Y.; Dong, Y.; Yang, J.; Zhang, L. Potential Pathogenic Bacteria in Seminal Microbiota of Patients with Different Types of Dysspermatism. Sci. Rep. 2020, 10, 6876. [Google Scholar] [CrossRef] [Green Version]
- Gachet, C.; Prat, M.; Burucoa, C.; Grivard, P.; Pichon, M. Spermatic Microbiome Characteristics in Infertile Patients: Impact on Sperm Count, Mobility, and Morphology. J. Clin. Med. 2022, 11, 1505. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Semen Parameter | Case Group (N = 13) | Control Group (N = 7) | Ref. Limit * |
|---|---|---|---|
| Viscosity | >2 cm filament 46% (6/13) | <2 cm filament 100% (7/7) | <2 cm filament |
| pH | 7.7 (7–8.2) | 7.6 (7.5–7.9) | ≥7.2 |
| Volume | 2.3 (1–4.4) | 3 (1.3–6.3) | ≥1.5 mL |
| Sperm concentration (×106 mL) | 20.2 (1.5–50) | 84.6 (35–170) | ≥15 × 106 mL |
| Total sperm number (×106 mL) | 40 (3.75–108) | 255 (45.5–418) | ≥39 × 106 ejaculate |
| Total sperm motility (PR + NP, %) | 29.5 (0–55) | 59.3 (40–80) | ≥40% |
| Progressive motility (PR, %) | 29.6 (0–65) | 63.6 (40–85) | ≥32% |
| Leucocytes (1 × 106/mL) | 2.7 (rare-10) | rare | <1 × 106 mL |
| Sperm morphology (%) | 3.4 (0–10%) | 6 (4–10) | ≥4% (normal forms) |
| Germinal cells | rare | rare | <10% |
| Agglutination | rare | absent | rare/absent |
| Rank | Taxon | p-Value | FDR |
|---|---|---|---|
| Order | Veillonellales_Selenomonadales | 2.8019 × 10−5 | 0.00098066 |
| Order | Peptostreptococcales_Tissierellales | 0.00013992 | 0.0024486 |
| Order | Pasteurellales | 0.0018522 | 0.021609 |
| Order | Actinomycetales | 0.005292 | 0.046305 |
| Order | Fusobacteriales | 0.0074557 | 0.047691 |
| Order | Campylobacterales | 0.0081756 | 0.047691 |
| Family | Peptoniphilus | 1.5837 × 10−5 | 0.00098188 |
| Family | Veillonellaceae | 0.00027895 | 0.0064464 |
| Family | Enterobacteriaceae | 0.00031192 | 0.0064464 |
| Family | Campylobacteraceae | 0.0015619 | 0.024209 |
| Family | Fusobacteriaceae | 0.0036006 | 0.039076 |
| Family | Pasteurellaceae | 0.0037816 | 0.039076 |
| Genus | Mannheimia | 6.6625 × 10−29 | 5.863 × 10−27 |
| Genus | Escherichia_Shigella | 0.00078541 | 0.034558 |
| Genus | Varibaculum | 0.0014709 | 0.043147 |
| Rank | Taxon | p-Value | FDR |
|---|---|---|---|
| Phylum | Actinobacteriota | 0.00139 | 0.00837 |
| Phylum | Bacteroidota | 0.00123 | 0.00837 |
| Phylum | Cyanobacteria | 0.000922 | 0.00837 |
| Class | Actinobacteria | 0.00145 | 0.00945 |
| Class | Alphaproteobacteria | 0.00111 | 0.00945 |
| Class | Bacteroidia | 0.00123 | 0.00945 |
| Class | Cyanobacteriia | 0.000922 | 0.00945 |
| Order | Burkholderiales | 0.000639 | 0.0144 |
| Order | Chitinophagales | 0.00133 | 0.0144 |
| Order | Chloroplast | 0.000922 | 0.0144 |
| Order | Corynebacteriales | 0.0015 | 0.0144 |
| Order | Micrococcales | 0.00101 | 0.0144 |
| Order | Propionibacteriales | 0.00034 | 0.0144 |
| Order | Rhizobiales | 0.00169 | 0.0144 |
| Order | Sphingomonadales | 0.00116 | 0.0144 |
| Order | Caulobacterales | 0.00217 | 0.0148 |
| Order | Staphylococcales | 0.00537 | 0.0304 |
| Order | Lachnospirales | 0.00747 | 0.0366 |
| Family | Alcaligenaceae | 0.00063 | 0.0118 |
| Family | Carnobacteriaceae | 0.000349 | 0.0118 |
| Family | Comamonadaceae | 0.0012 | 0.0118 |
| Family | Gemellaceae | 0.000764 | 0.0118 |
| Family | Micrococcaceae | 0.000663 | 0.0118 |
| Family | Neisseriaceae | 0.000173 | 0.0118 |
| Family | Propionibacteriaceae | 0.00034 | 0.0118 |
| Family | Sphingomonadaceae | 0.00116 | 0.0118 |
| Family | Xanthobacteraceae | 0.00107 | 0.0118 |
| Family | Yersiniaceae | 0.00041 | 0.0118 |
| Family | Chitinophagaceae | 0.00133 | 0.0121 |
| Family | Nocardiaceae | 0.00184 | 0.0144 |
| Family | Caulobacteraceae | 0.00217 | 0.016 |
| Family | Beijerinckiaceae | 0.00247 | 0.0168 |
| Family | Corynebacteriaceae | 0.00256 | 0.0168 |
| Family | Leptotrichiaceae | 0.00276 | 0.0171 |
| Family | Burkholderiaceae | 0.00415 | 0.0245 |
| Family | Staphylococcaceae | 0.00611 | 0.0343 |
| Family | Lachnospiraceae | 0.00747 | 0.0372 |
| Family | Streptococcaceae | 0.00758 | 0.0372 |
| Genus | Neisseria | 5.28 × 10−5 | 0.00887 |
| Genus | Acidipropionibacterium | 0.000207 | 0.0115 |
| Genus | Cutibacterium | 0.000302 | 0.0115 |
| Genus | Granulicatella | 0.000412 | 0.0115 |
| Genus | Serratia | 0.00041 | 0.0115 |
| Genus | Kocuria | 0.000519 | 0.0124 |
| Genus | Achromobacter | 0.00063 | 0.0128 |
| Genus | Gemella | 0.000764 | 0.0128 |
| Genus | Bradyrhizobium | 0.00107 | 0.0164 |
| Genus | Sphingomonas | 0.00123 | 0.0173 |
| Genus | Sediminibacterium | 0.00146 | 0.0188 |
| Genus | Gordonia | 0.00184 | 0.022 |
| Genus | Leptotrichia | 0.00276 | 0.0273 |
| Genus | Methylobacterium_Methylorubrum | 0.00247 | 0.0273 |
| Genus | Lautropia | 0.00415 | 0.0387 |
| Genus | Corynebacterium | 0.00472 | 0.0405 |
| Genus | Lactococcus | 0.00482 | 0.0405 |
| Genus | Actinomyces | 0.00649 | 0.0452 |
| Genus | Caulobacter | 0.00659 | 0.0452 |
| Genus | Lawsonella | 0.00673 | 0.0452 |
| Genus | Staphylococcus | 0.00599 | 0.0452 |
| Genus | Streptococcus | 0.00771 | 0.0498 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veneruso, I.; Cariati, F.; Alviggi, C.; Pastore, L.; Tomaiuolo, R.; D’Argenio, V. Metagenomics Reveals Specific Microbial Features in Males with Semen Alterations. Genes 2023, 14, 1228. https://doi.org/10.3390/genes14061228
Veneruso I, Cariati F, Alviggi C, Pastore L, Tomaiuolo R, D’Argenio V. Metagenomics Reveals Specific Microbial Features in Males with Semen Alterations. Genes. 2023; 14(6):1228. https://doi.org/10.3390/genes14061228
Chicago/Turabian StyleVeneruso, Iolanda, Federica Cariati, Carlo Alviggi, Lucio Pastore, Rossella Tomaiuolo, and Valeria D’Argenio. 2023. "Metagenomics Reveals Specific Microbial Features in Males with Semen Alterations" Genes 14, no. 6: 1228. https://doi.org/10.3390/genes14061228
APA StyleVeneruso, I., Cariati, F., Alviggi, C., Pastore, L., Tomaiuolo, R., & D’Argenio, V. (2023). Metagenomics Reveals Specific Microbial Features in Males with Semen Alterations. Genes, 14(6), 1228. https://doi.org/10.3390/genes14061228