
A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Genome Size Estimation and Genome Assembly
2.3. Hi-C-assisted Chromosome-Level Assembly
2.4. Repeat Annotation, Gene Prediction, and Gene Functional Annotation
2.5. Comparative Genomic Analyses and Selection Analysis
3. Results and Discussion
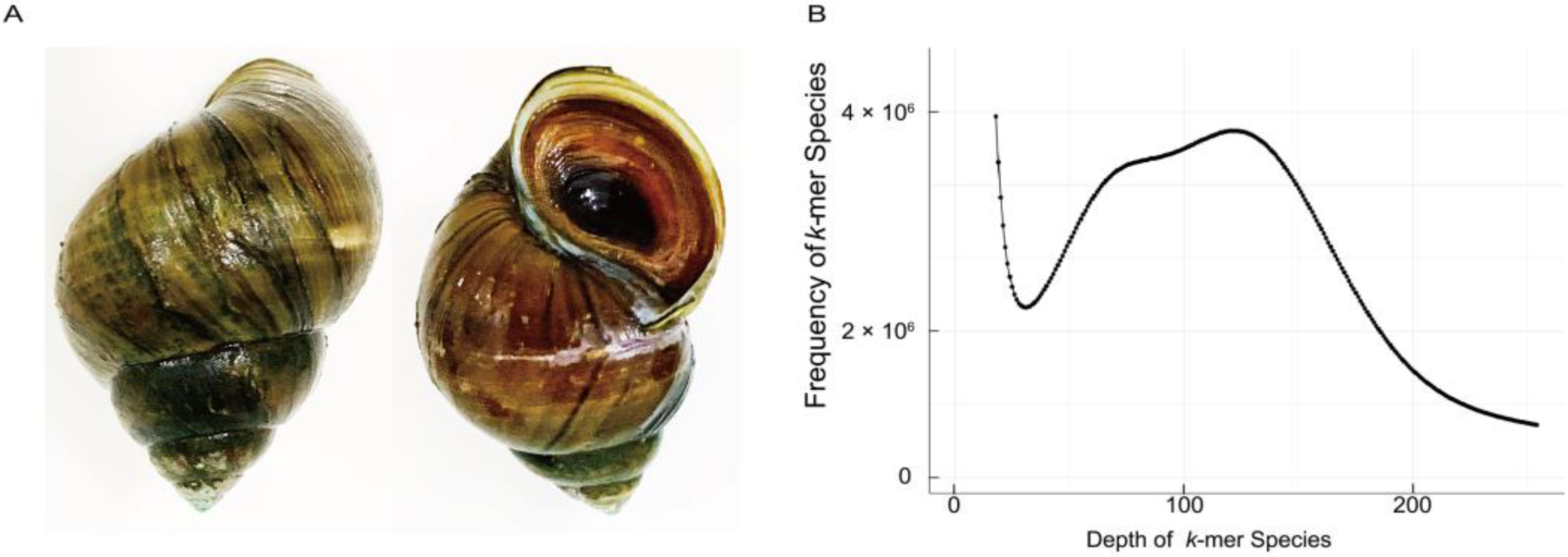
3.1. Initial Characterization of C. cathayensis Genome
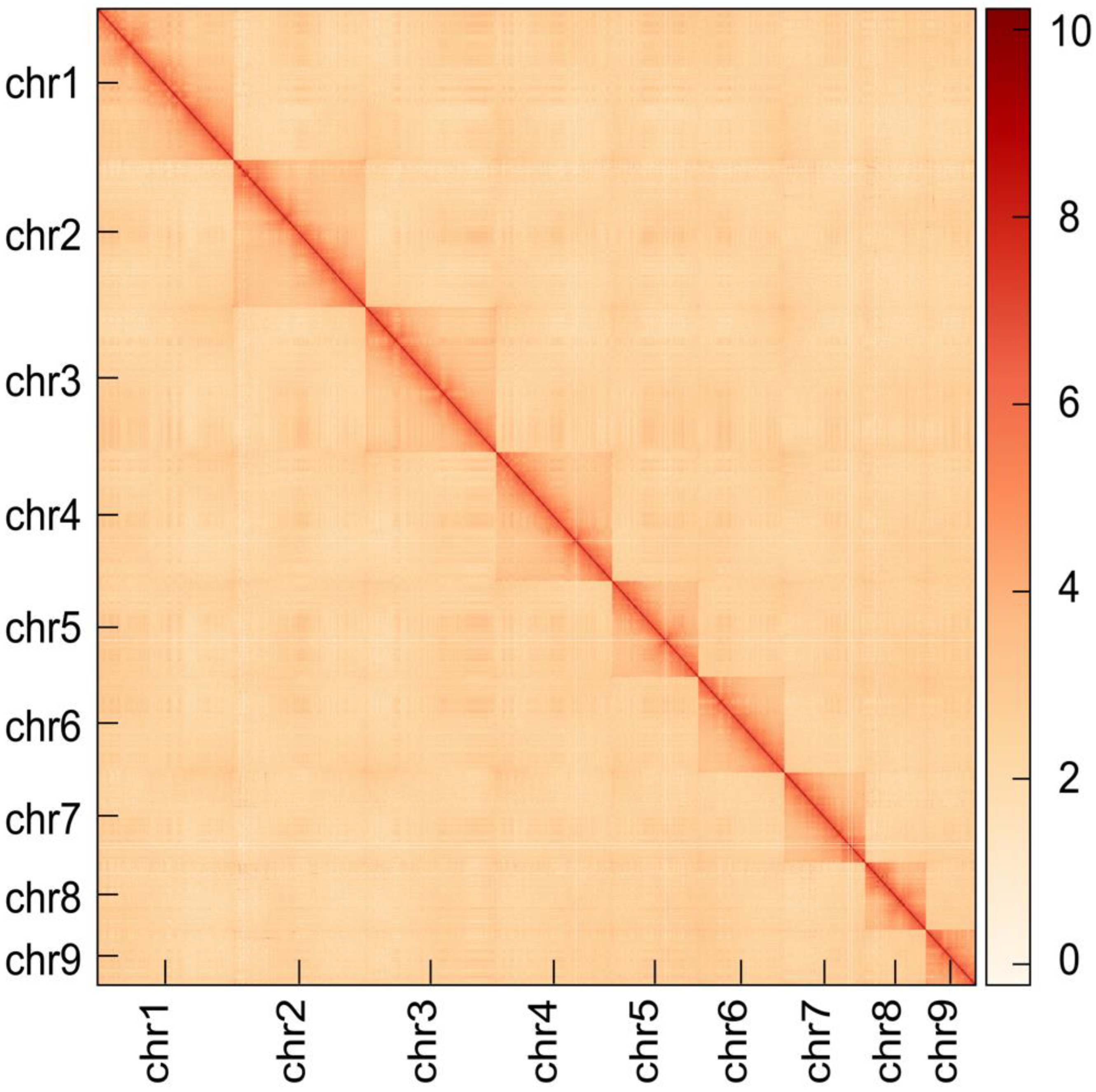
3.2. Genome Assembly and Assessment
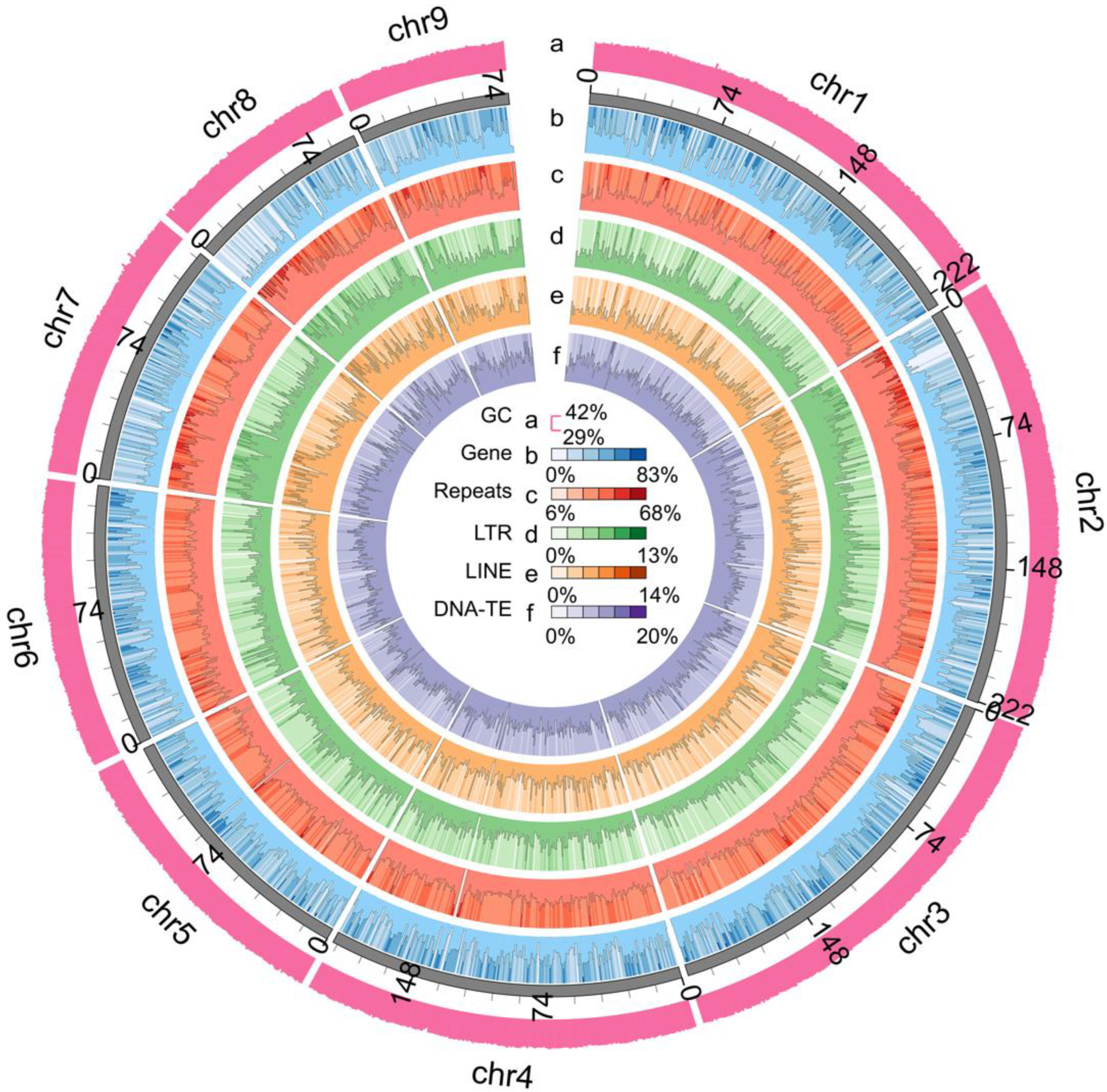
3.3. Gene Structure and Function Annotations
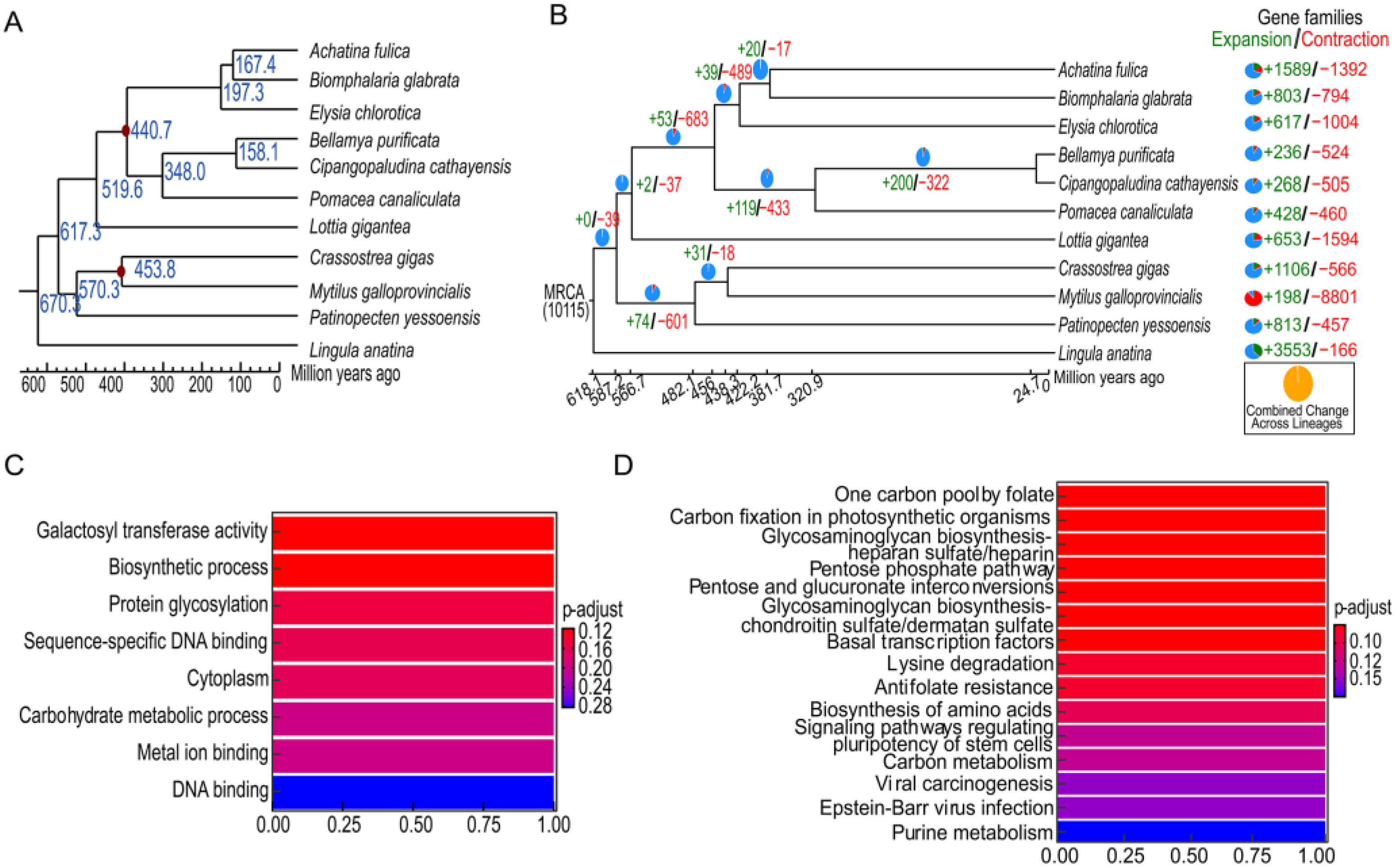
3.4. Comparative Genomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, H.; Zhang, J.E.; Luo, H.; Luo, M.; Guo, J.; Deng, Z.; Zhao, B. The complete mitochondrial genome of the mudsnail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 1892–1894. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, W.; Wang, Y.; Wang, E. Economic Fauna Sinica of China, Freshwater Mollusks; Science Press: Beijing, China, 1979. [Google Scholar]
- Wang, J.G.; Zhang, D.; Jakovlić, I.; Wang, W.M. Sequencing of the complete mitochondrial genomes of eight freshwater snail species exposes pervasive paraphyly within the Viviparidae family (Caenogastropoda). PLoS ONE 2017, 12, e0181699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Cao, J.Y.; Ma, C.; Ma, X.Y.; Lv, G.H.; Wen, H.B. Genetic diversity and genetic differentiation analysis of Bellamya purificata in eleven populations based on the microsatellite makers. Freshw. Fish. 2022, 52, 16–21. (In Chinese) [Google Scholar] [CrossRef]
- Huang, S.Q.; Jiang, H.J.; Zhang, L.; Gu, Q.H.; Wang, W.M.; Wen, Y.H.; Luo, F.; Jin, W.; Cao, X. Integrated proteomic and transcriptomic analysis reveals that polymorphic shell colors vary with melanin synthesis in Bellamya purificata snail. J. Proteom. 2021, 230, 103950. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Cao, X.J.; Ma, X.Y.; Lv, G.H.; Xu, G.C.; Xu, P.; Sun, B.; Xu, D.P.; Wen, H.B. Chromosome-level genome assembly of the freshwater snail Bellamya purificata (Caenogastropoda). Zool. Res. 2022, 43, 683–686. [Google Scholar] [CrossRef]
- Jiang, J.Y.; Li, W.H.; Wu, Y.Y.; Cheng, C.X.; Ye, Q.Q.; Feng, J.X.; Xie, Z.X. Effects of cadmium exposure on intestinal microflora of Cipangopaludina cathayensis. Front Microbiol. 2022, 13, 984757. [Google Scholar] [CrossRef]
- Zhang, G.; Yin, D.; He, T.; Xu, Y.; Ran, S.; Zhou, X.; Tian, X.; Wang, Y. Mercury Bioaccumulation in Freshwater Snails as Influenced by Soil Composition. Bull. Environ. Contam. Toxicol. 2021, 106, 153–159. [Google Scholar] [CrossRef]
- Guo, Z.F.; Ai, H. Research progress of Chinese Cipangopaludina cathayensis and its bioactive constituent. Food Res. Dev. 2015, 36, 132–134. (In Chinese) [Google Scholar] [CrossRef]
- Bhattacharya, S.; Chakraborty, M.; Bose, M.; Mukherjee, D.; Roychoudhury, A.; Dhar, P.; Mishra, R. Indian freshwater edible snail Bellamya bengalensis lipid extract prevents T cell mediated hypersensitivity and inhibits LPS induced macrophage activation. J. Ethnopharmacol. 2014, 157, 320–329. [Google Scholar] [CrossRef]
- Wang, C.; Liu, J.; Huang, Y.; Zhang, X. In vitro polysaccharide extraction from Cipangopaludina cathayensis and its pharmacological potential. J. Environ. Biol. 2016, 37, 1069–1072. [Google Scholar]
- Zhao, T.; Xiong, J.; Chen, W.; Xu, A.; Zhu, D.; Liu, J. Purification and Characterization of a Novel Fibrinolytic Enzyme from Cipangopaludina Cahayensis. Iran. J. Biotechnol. 2021, 19, e2805. [Google Scholar] [CrossRef]
- Dhiman, V.; Pant, D. Human health and snails. J. Immunoass. Immunochem. 2021, 42, 211–235. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Cheng, C.X.; Yang, L.; Ye, Q.Q.; Li, W.H.; Jiang, J.Y. Characterization of Gut Microbiome in the Mud Snail Cipangopaludina cathayensis in Response to High-Temperature Stress. Animals 2022, 12, 2361. [Google Scholar] [CrossRef]
- Li, K.; Jiang, W.; Hui, Y.; Kong, M.; Feng, L.Y.; Gao, L.Z.; Li, P.; Lu, S. Gapless indica rice genome reveals synergistic contributions of active transposable elements and segmental duplications to rice genome evolution. Mol. Plant 2021, 14, 1745–1756. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Roach, M.J.; Schmidt, S.A.; Borneman, A.R. Purge Haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 2018, 19, 460. [Google Scholar] [CrossRef]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 4, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, W265–W268. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21 (Suppl. 1), i152–i158. [Google Scholar] [CrossRef]
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. 1), i351–i358. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Doerks, T.; Copley, R.R.; Schultz, J.; Ponting, C.P.; Bork, P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res. 2002, 12, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simakov, O.; Marletaz, F.; Cho, S.J.; Edsinger-Gonzales, E.; Havlak, P.; Hellsten, U.; Kuo, D.H.; Larsson, T.; Lv, J.; Arendt, D.; et al. Insights into bilaterian evolution from three spiralian genomes. Nature 2013, 493, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adema, C.M.; Hillier, L.W.; Jones, C.S.; Loker, E.S.; Knight, M.; Minx, P.; Oliveira, G.; Raghavan, N.; Shedlock, A.; Do Amaral, L.R.; et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat. Commun. 2017, 8, 15451. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Liu, C.; Zhang, Y.; Ren, Y.; Wang, H.; Li, S.; Jiang, F.; Yin, L.; Qiao, X.; Zhang, G.; et al. The genome of the golden apple snail Pomacea canaliculata provides insight into stress tolerance and invasive adaptation. GigaScience 2018, 7, giy101. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Liu, Q.; Huang, Y.; Mao, G.; Yue, Z.; Abe, E.M.; Li, J.; Wu, Z.; Li, S.; et al. A chromosomal-level genome assembly for the giant African snail Achatina fulica. Gigascience 2019, 8, giz124. [Google Scholar] [CrossRef]
- Zhao, D.; Zhao, M.; Wu, Z.D. The karyotype of five species of freshwater snails of the family Viviparidae. Acta Zool. Sin. 1988, 4, 364–370. [Google Scholar]
- Calvete, O.; Reyes, J.; Valdés-Socin, H.; Martin, P.; Marazuela, M.; Barroso, A.; Escalada, J.; Castells, A.; Torres-Ruiz, R.; Rodríguez-Perales, S.; et al. Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid-Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario. Cells 2021, 10, 3500. [Google Scholar] [CrossRef]
- Nikpour, M.; Noborn, F.; Nilsson, J.; Van Damme, T.; Kaye, O.; Syx, D.; Malfait, F.; Larson, G. Glycosaminoglycan linkage region of urinary bikunin as a potentially useful biomarker for β3GalT6-deficient spondylodysplastic Ehlers-Danlos syndrome. JIMD Rep. 2022, 63, 462–467. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, B.; Jin, W.; Fu, H.; Sun, B.; Yang, S.; Ma, X.; Wen, H.; Wu, X.; Wang, H.; Cao, X. A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Genes 2023, 14, 1365. https://doi.org/10.3390/genes14071365
Ma B, Jin W, Fu H, Sun B, Yang S, Ma X, Wen H, Wu X, Wang H, Cao X. A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Genes. 2023; 14(7):1365. https://doi.org/10.3390/genes14071365
Chicago/Turabian StyleMa, Benhe, Wu Jin, Huiyun Fu, Bing Sun, Su Yang, Xueyan Ma, Haibo Wen, Xiaoping Wu, Haihua Wang, and Xiaojuan Cao. 2023. "A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae)" Genes 14, no. 7: 1365. https://doi.org/10.3390/genes14071365
APA StyleMa, B., Jin, W., Fu, H., Sun, B., Yang, S., Ma, X., Wen, H., Wu, X., Wang, H., & Cao, X. (2023). A High-Quality Chromosome-Level Genome Assembly of a Snail Cipangopaludina cathayensis (Gastropoda: Viviparidae). Genes, 14(7), 1365. https://doi.org/10.3390/genes14071365