Genetic Evaluation of Natural Populations of the Endangered Conifer Thuja koraiensis Using Microsatellite Markers by Restriction-Associated DNA Sequencing


Abstract
:1. Introduction
2. Materials and Methods
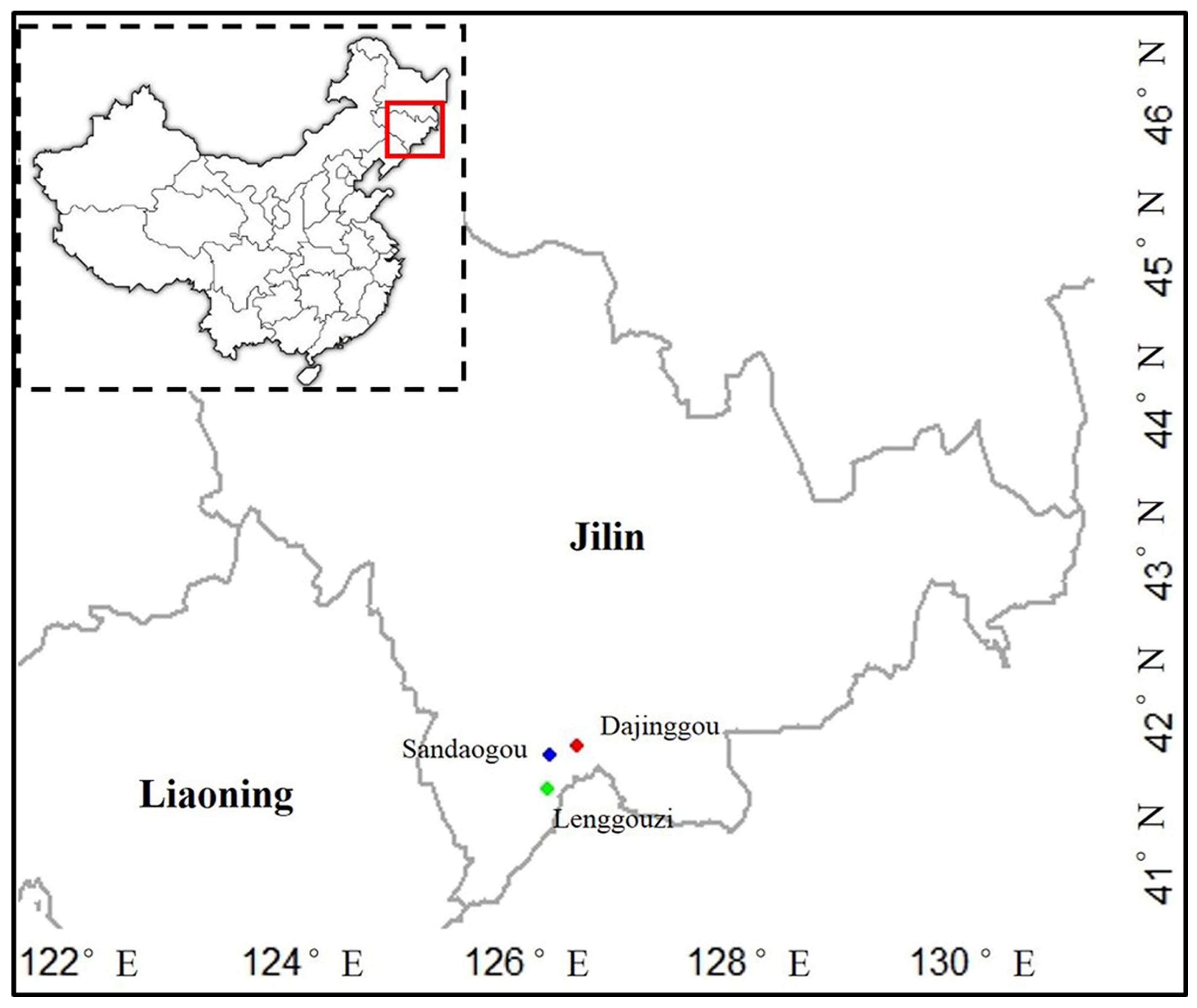
2.1. Plant Material and DNA Extraction
2.2. RAD Library Preparation, Sequencing, and Assembly
2.3. RAD Sequence Analysis
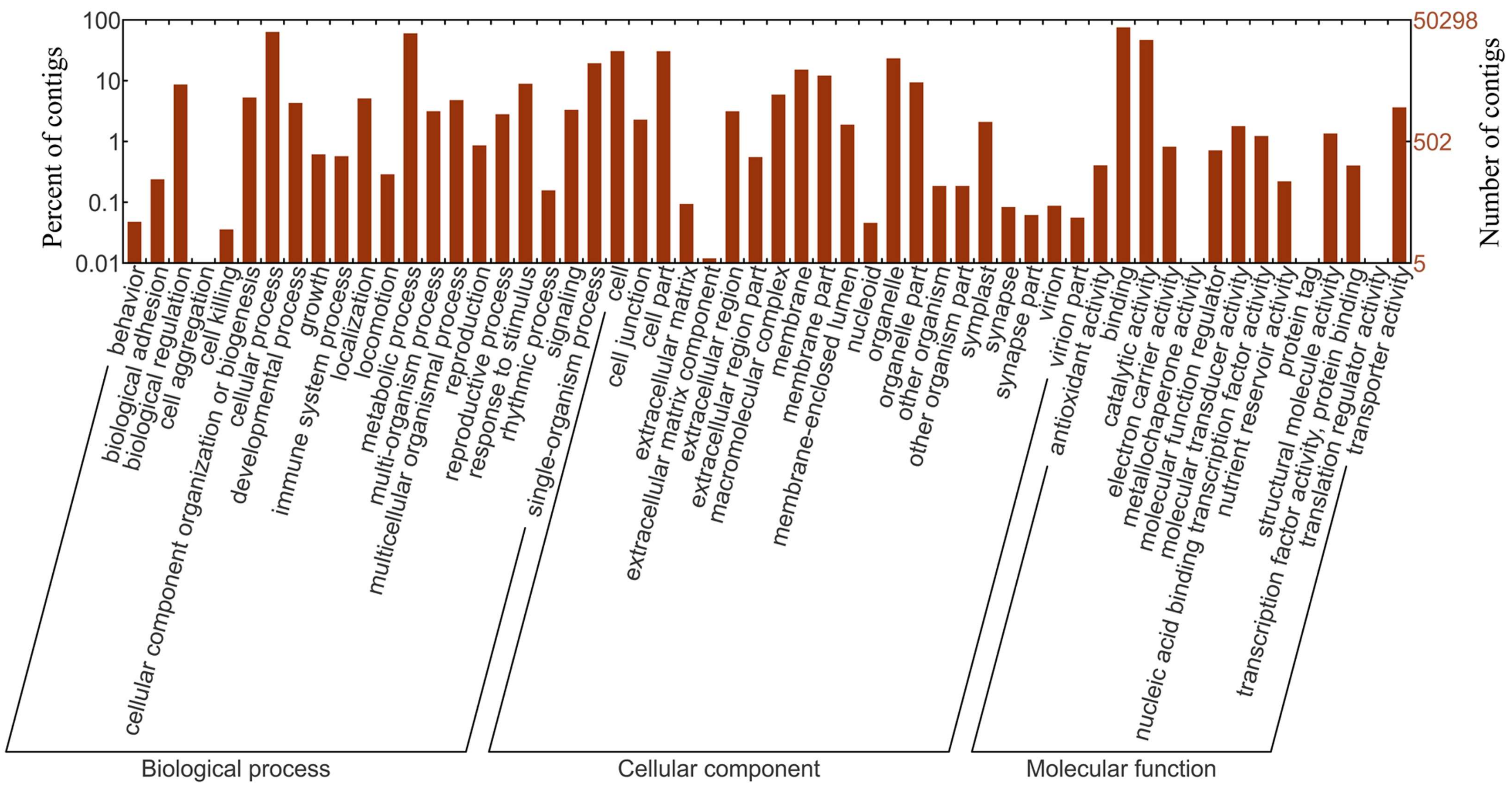
2.4. Sequence Annotation and Classification
2.5. SSR Identification and Marker Design
2.6. Data Analysis
3. Results
3.1. Sequencing, Contigs Assembly and Functional Annotation
3.2. Frequency and Distribution of SSR Markers
3.3. Development, Validation and Polymorphism of SSR Markers
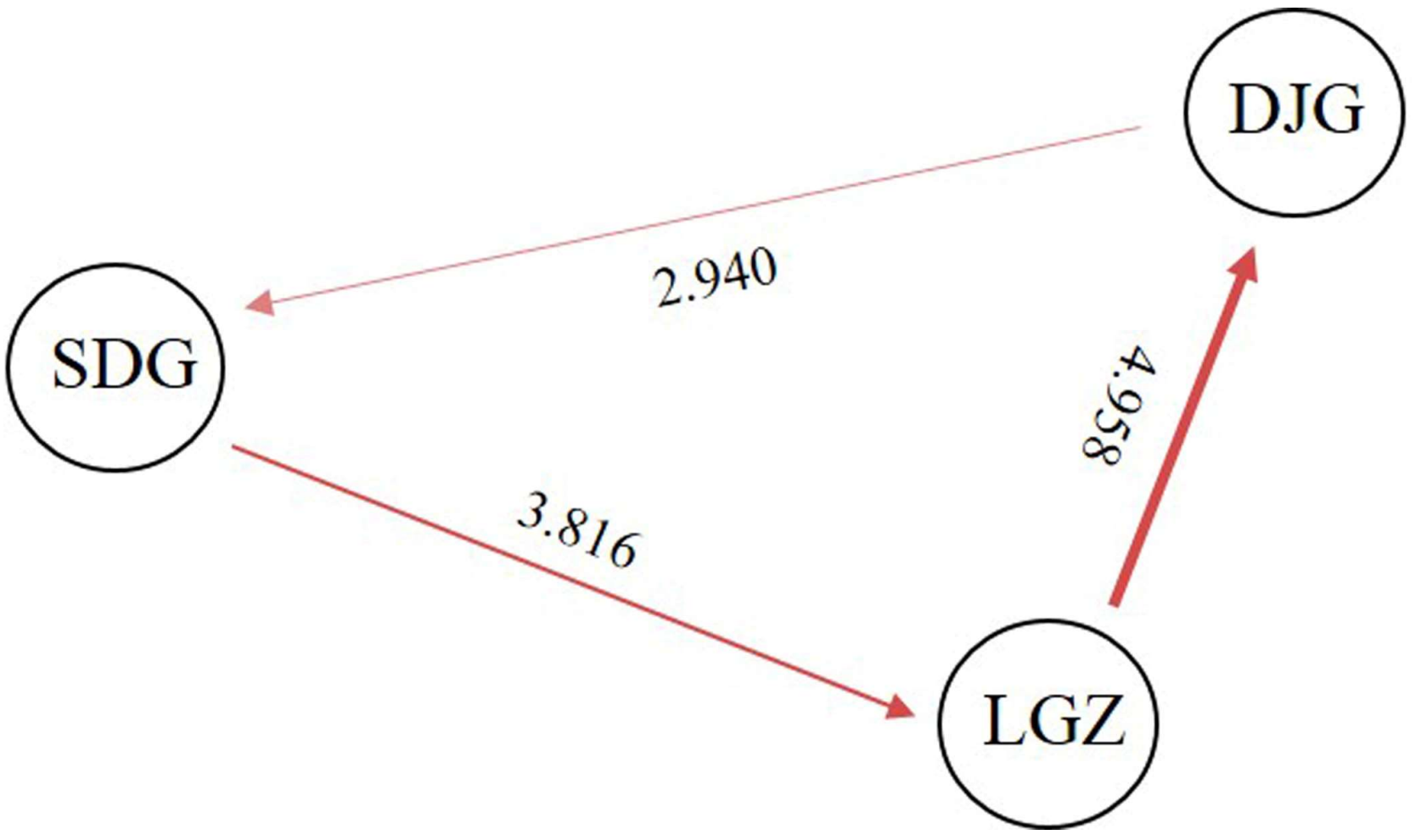
3.4. Population Genetic Diversity and Differentiation
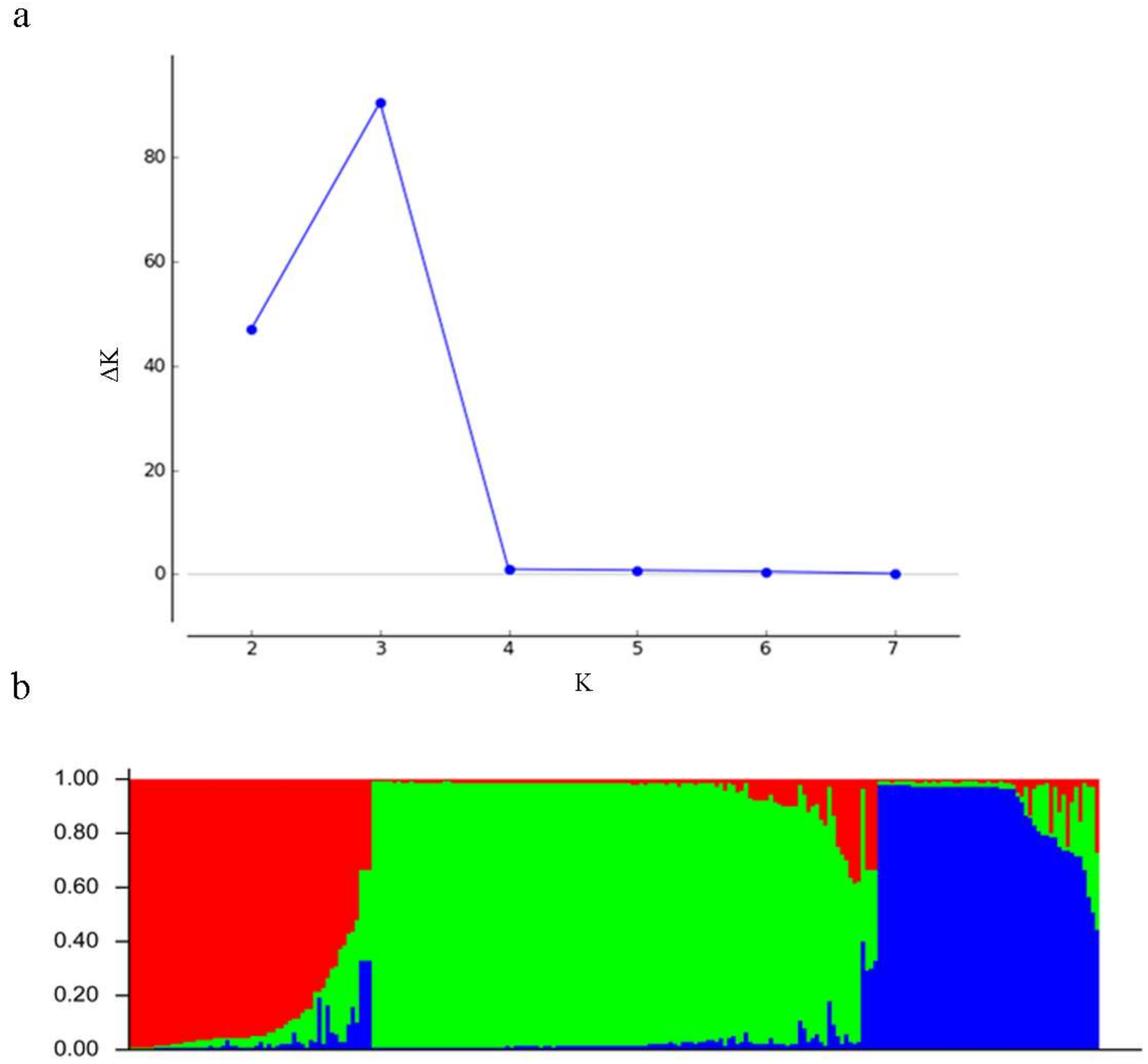
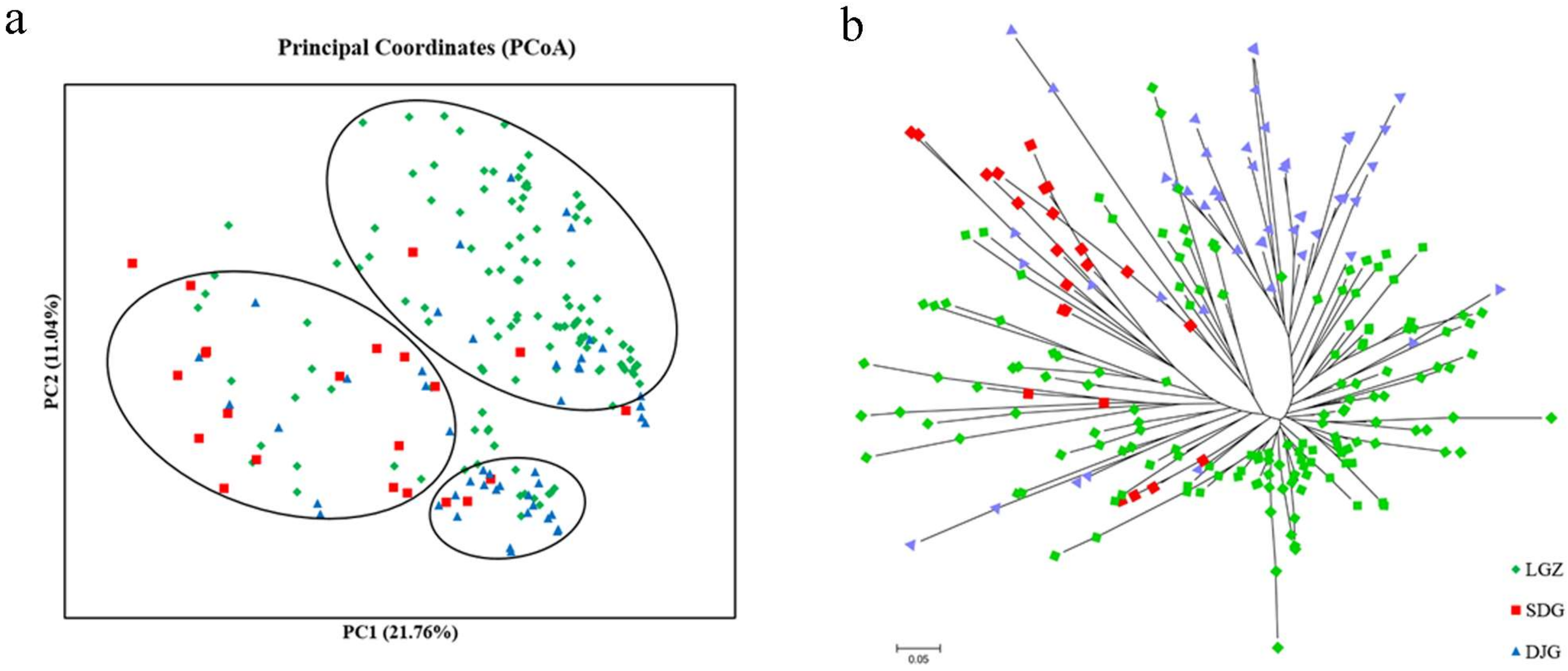
3.5. Population Genetic Structure
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sun, B.; Cui, Y.M.; Wang, H.F.; Ferguson, D.K.; Xiang, Q.P.; Ma, Q.W.; Wang, Y.F. Recognizing the species of Thuja (Cupressaceae) based on their cone and foliage morphology. Phytotaxa 2015, 219, 101–117. [Google Scholar] [CrossRef]
- Yin, H.; Jin, H.; Zhao, Y.; Qin, L.W.; Dai, Y.H.; Liu, L.; Zhao, W. Present situation and conservation strategy of rare and endangered species Thuja koraiensis in Changbai mountain. J. Beihua Univ. 2016, 17, 40–42. [Google Scholar]
- Fu, L.G. China Plant Red Data Book; Science Press: Beijing, China, 1992; pp. 38–40. ISBN 7-03-000485-X. [Google Scholar]
- He, X.; Lin, J.; Hu, Y.; Wang, X.; Li, F. Comparison among threatened categories of conifers from China. Chin. Biodivers. 1996, 4, 45–51. [Google Scholar]
- Liu, C.; Wang, Y.; Shi, S. Height growth regularity of Thuja koraiensis. Jilin For. Sci. Technol. 2009, 38, 20–22. [Google Scholar]
- Chung, I.M.; Praveen, N.; Ahmad, A. Composition of the essential oil and antioxidant activity of petroleum ether extract of Thuja koraiensis. Asian J. Chem. 2011, 23, 3703–3706. [Google Scholar]
- Tang, X.; Li, Z.Y.; Hu, Y.X. Wood anatomy of Thuja sutchuenensis endemic to China. J. Wuhan Bot. Res. 2005, 23, 149–153. [Google Scholar]
- Wang, H.S.; Deng, Z.G.; Huang, Z.Z.; Li, R.X. Structural research on secondary xylem of stem of Thuja koraiensis Nakai under SEM. J. Tonghua Norm. Univ. 2007, 28, 22–24. [Google Scholar]
- Yin, H.; Zhao, Y.; Cui, K.; Liu, L.J.; Yu, C.; Chen, Q.; Dai, Y.; Zhao, W. Asexual reproduction technique of Thuja koraiensis Nakai. Chin. Wild Plant Resour. 2013, 32, 68–69. [Google Scholar]
- Yang, Z.; Tian, Z.; Liu, Q.; Sun, Y. Studies on the chemical constituents of the volatile oil from leaves of Thuja Koraiensis Nakai maxim. J. Northeast Norm. Univ. 1994, 1, 136–140. [Google Scholar]
- Qi, J.Z.; Sun, G.R.; Yang, W.S.; Sun, R.C.; Xue, F. Analysis on the chemical constituents of essential oil from branches and leaves of Thuja Koraiensis Nakai. J. Plant Resour. Environ. 1995, 4, 61–62. [Google Scholar]
- Cohen, J.I.; Williams, J.T.; Plucknett, D.L.; Shands, H. Ex situ conservation of plant genetic resources: Global development and environmental concerns. Science 1991, 253, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Ouborg, N.J. Integrating population genetics and conservation biology in the era of genomics. Biol. Lett. 2010, 6, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Aranzana, M.; Carbó, J.; Arús, P. Microsatellite variability in peach [Prunus persica (L.) Batsch]: Cultivar identification, marker mutation, pedigree inferences and population structure. Theor. Appl. Genet. 2003, 106, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, V.; Nepolean, T.; Senthilvel, S.; Varshney, R.K.; Vadez, V.; Srivastava, R.K.; Shah, T.M.; Supriya, A.; Kumar, S.; Kumari, B.R.; et al. Pearl millet (Pennisetum glaucum (L.) R. Br.) consensus linkage map constructed using four RIL mapping populations and newly developed EST-SSRs. BMC Genom. 2013, 14, 159. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.M.H.; Fowler, J.C.S.; Thomas, M.R. An evaluation of sequence tagged microsatellite site markers for genetic analysis within Citrus and related species. Genome 1995, 38, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Ren, X.L.; Liu, W.Z. Genetic diversity of SSR markers in wild populations of Tapiscia sinensis, an endangered tree species. Biochem. Syst. Ecol. 2016, 69, 1–5. [Google Scholar] [CrossRef]
- Szczecińska, M.; Sramko, G.; Wołosz, K.; Sawicki, J. Genetic diversity and population structure of the rare and endangered plant species Pulsatilla patens (L.) Mill in East Central Europe. PLoS ONE 2016, 11, e0151730. [Google Scholar]
- Forrest, A.; Escudero, M.; Heuertz, M.; Wilson, Y.; Cano, E.; Vargas, P. Testing the hypothesis of low genetic diversity and population structure in narrow endemic species: The endangered Antirrhinum charidemi (Plantaginaceae). Bot. J. Linn. Soc. 2017, 183, 260–270. [Google Scholar] [CrossRef]
- Aboukhalid, K.; Machon, N.; Lambourdière, J.; Abdelkrim, J.; Bakha, M.; Douaik, A.; Korbecka-Glinkaf, G.; Gabouna, F.; Tomig, F.; Lamirib, A.; et al. Analysis of genetic diversity and population structure of the endangered Origanum compactum from Morocco, using SSR markers: Implication for conservation. Biol. Conserv. 2017, 212, 172–182. [Google Scholar] [CrossRef]
- Ma, Q.; Feng, K.; Yang, W.; Chen, Y.; Yu, F.; Yin, T. Identification and characterization of nucleotide variations in the genome of Ziziphus jujuba (Rhamnaceae) by next generation sequencing. Mol. Biol. Rep. 2014, 41, 3219–3223. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.M.; Toledo-Silva, G.; Cardoso-Silva, C.B.; Da Silva, C.C.; de Araujo Andreotti, I.A.; Conson, A.R.O.; Mantello, C.C.; Guen, V.; de Souza, A.P. Development of single nucleotide polymorphism markers in the large and complex rubber tree genome using next-generation sequence data. Mol. Breed. 2016, 36, 1–10. [Google Scholar] [CrossRef]
- Motalebipour, E.Z.; Kafkas, S.; Khodaeiaminjan, M.; Çoban, N.; Gözel, H. Genome survey of pistachio (Pistacia vera L.) by next generation sequencing: Development of novel SSR markers and genetic diversity in Pistacia species. BMC Genom. 2016, 17, 998. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Barchi, L.; Lanteri, S.; Portis, E.; Acquadro, A.; Valè, G.; Toppino, L.; Rotino, G.L. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genom. 2011, 12, 304. [Google Scholar] [CrossRef] [PubMed]
- Vandepitte, K.; Honnay, O.; Mergeay, J.; Breyne, P.; Roldán-Ruiz, I.; De Meyer, T. SNP discovery using Paired-End RAD-tag sequencing on pooled genomic DNA of Sisymbrium austriacum (Brassicaceae). Mol. Ecol. Resour. 2013, 13, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, X.; Zhang, B.; Shen, C.; Lin, Z. Enrichment of an intraspecific genetic map of upland cotton by developing markers using parental RAD sequencing. DNA Res. 2015, 22, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Baek, J.; Carrasquilla-Garcia, N.; Penmetsa, R.V. Genome-wide polymorphism detection in peanut using next-generation restriction-site-associated DNA (RAD) sequencing. Mol. Breed. 2015, 35, 145. [Google Scholar] [CrossRef]
- Tian, Z.; Zhang, F.; Liu, H.; Gao, Q.; Chen, S. Development of SSR markers for a Tibetan medicinal plant, Lancea tibetica (Phrymaceae), based on RAD sequencing. Appl. Plant Sci. 2016, 4, 1600076. [Google Scholar] [CrossRef] [PubMed]
- Durand, J.; Bodénès, C.; Chancerel, E.; Frigerio, J.M.; Vendramin, G.; Sebastiani, F.; Buonamici, A.; Gailing, O.; Koelewijn, H.P.; Villani, F.; et al. A fast and cost-effective approach to develop and map EST-SSR markers: Oak as a case study. BMC Genom. 2010, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lesser, M.R.; Parchman, T.L.; Buerkle, C. Cross-species transferability of SSR loci developed from transcriptome sequencing in lodgepole pine. Mol. Ecol. Resour. 2012, 12, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Chutimanitsakun, Y.; Nipper, R.W.; Cuesta-Marcos, A.; Cistué, L.; Corey, A.; Filichkina, T.; Johnson, E.A.; Hayes, P.M. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genom. 2011, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.J.; Ma, Y.P.; Wang, S.; Hu, X.G.; Huang, L.S.; Li, Y.; Wang, X.R.; Mao, J.F. Genetic evaluation of the breeding population of a valuable reforestation conifer Platycladus orientalis (Cupressaceae). Sci. Rep. 2016, 6, 34821. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Cui, B.B.; Mao, J.F.; Pang, X.M.; Li, Y.Y. Novel polymorphic EST-derived microsatellite markers for the red-listed five needle pine, Pinus dabeshanensis. Conserv. Genet. Resour. 2015, 7, 191–192. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete reimplementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Li, C.Y.; Xiong, F. Isolation and characterization of 19 polymorphic microsatellite loci from Neosalanx taihuensis, a rapidly invasive and adaptative species. Biochem. Syst. Ecol. 2008, 61, 121–123. [Google Scholar] [CrossRef]
- Beerli, P.; Palczewski, M. Unified framework to evaluate Panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F.; Tateno, Y. Accuracy of estimated phylogenetic trees from molecular data. J. Mol. Evol. 1983, 19, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S. PowerMarker: New genetic data analysis software. Version 3.23. Bioinformatics 2004, 9, 2128–2129. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Yang, Y.; Wang, Z.; Qi, B.; Yin, Y.; Li, H. Development and characterization of EST-SSR markers in Taxodium ‘zhongshansa’. Plant Mol. Biol. Report. 2015, 33, 1–11. [Google Scholar] [CrossRef]
- Scaglione, D.; Acquadro, A.; Portis, E.; Tirone, M.; Knapp, S.J.; Lanteri, S. RAD tag sequencing as a source of SNP markers in Cynara cardunculus L. BMC Genom. 2012, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Pegadaraju, V.; Nipper, R.; Hulke, B.; Qi, L.; Schultz, Q. De novo sequencing of sunflower genome for SNP discovery using RAD (restriction site associated DNA) approach. BMC Genom. 2013, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Zhao, L.; Eaton, D.A.R.; Li, D.Z.; Guo, Z.H. Identification of SNP markers for inferring phylogeny in temperate bamboos (Poaceae: Bambusoideae) using RAD sequencing. Mol. Ecol. Resour. 2013, 13, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Xiao, G.; Guo, J.; Fei, Z.; Xu, Y.; Roe, B.A.; Wang, Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genom. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.G.; Liu, H.; Zhang, J.Q.; Sun, Y.Q.; Jin, Y.Q.; Zhao, W.; El-Kassaby, Y.A.; Wang, X.R.; Mao, J.F. Global transcriptome analysis of Sabina chinensis (Cupressaceae), a valuable reforestation conifer. Mol. Breed. 2016, 36, 99. [Google Scholar] [CrossRef]
- Jaillon, O.; Aury, J.; Noel, B.; Policriti, A.; Clepet, C.; Casagrande, A.; Choisne, N.; Aubourg, S.; Vitulo, N.; Jubin, C.; et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 2007, 449, 463–467. [Google Scholar] [PubMed]
- Niwa, Y.; Yamashino, T.; Mizuno, T. Circadian clock regulates photoperiodic response of hypocotyl elongation through a coincidence mechanism in Arabidopsis thaliana. Plant Cell Physiol. 2009, 50, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.G.; Liu, H.; Jin, Y.; Sun, Y.Q.; Li, Y.; Zhao, W.; El-Kassaby, Y.A.; Wang, X.R.; Mao, J.F. De novo transcriptome assembly and characterization for the widespread and stress-tolerant conifer Platycladus orientalis. PLoS ONE 2016, 11, e0148985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, B.; Li, H.; Chang, Y.; Han, Y.; Li, J.; Wei, G.; Zhao, S.; Khan, M.; Zhou, Y.; et al. Identification, characterization, and utilization of genome-wide simple sequence repeats to identify a QTL for acidity in apple. BMC Genom. 2012, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.M.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S.; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genom. 2010, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Lu, C.; Zhang, Y.; Song, G. Distribution and characterization of simple sequence repeats in Gossypium raimondii genome. Bioinformation 2012, 8, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Elbaidouri, M.; Abernathy, B.; Chen, H.L.; Wang, S.H.; Lee, S.H.; Jackson, S.A.; Cheng, X.Z. Distribution and analysis of SSR in mung bean (Vigna radiata, L.) genome based on an SSR-enriched library. Mol. Breed. 2015, 35, 25. [Google Scholar] [CrossRef]
- Kumpatla, S.P. Computational Mining and Survey of Simple Sequence Repeats (SSRs) in Expressed Sequence Tags (ESTs) of Dicotyledonous Plants. Master’s Thesis, Indiana University-Purdue University, Indianapolis, IN, USA, 2004. [Google Scholar]
- Sonah, H.; Deshmukh, R.K.; Sharma, A.; Singh, V.P.; Gupta, D.K.; Gacche, R.N.; Rana, J.C.; Singh, N.K.; Sharma, T.R. Genome-wide distribution and organization of microsatellites in plants: An insight into marker development in Brachypodium. PLoS ONE 2011, 6, e21298. [Google Scholar] [CrossRef] [PubMed]
- Kumpatla, S.; Mukhopadhyay, S. Mining and survey of simple sequence repeats in expressed sequence tags of dicotyledonous species. Genome 2005, 48, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Yang, H.; Sun, P.; Li, J.; Zhang, J.; Guo, Y.; Han, X.; Zhang, G.; Lu, M.; Hu, J. De novo transcriptome assembly, development of EST-SSR markers and population genetic analyses for the desert biomass willow, Salix psammophila. Sci. Rep. 2016, 6, 39591. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genome, survey and analysis. Genome Res. 2000, 10, 1967–1981. [Google Scholar] [CrossRef]
- Pandey, M.; Rajora, O.P. Genetic diversity and differentiation of core vs. peripheral populations of eastern white cedar, Thuja occidentalis (Cupressaceae). Am. J. Bot. 2012, 99, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Iwaizumi, M.G.; Tsuda, Y.; Ohtani, M.; Tsumura, Y.; Takahashi, M. Recent distribution changes affect geographic clines in genetic diversity and structure of Pinus densiflora natural populations in Japan. For. Ecol. Manag. 2013, 304, 407–416. [Google Scholar] [CrossRef]
- Lin, Z.; Lou, A. Old-Growth Platycladus orientalis as a resource for reproductive capacity and genetic diversity. Plos ONE 2013, 8, e56489. [Google Scholar]
- Huh, M.; Huh, H.W. Patterns of genetic diversity and population structure of the clonal herb, Potentilla fragarioides var. sprengeliana (Rosaceae) in Korea. Acta Bot. Sin. 1999, 42, 64–70. [Google Scholar]
- Cole, C.T. Genetic variation in rare and common plants. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 213–237. [Google Scholar] [CrossRef]
- Xu, X.X.; Cheng, F.Y.; Xian, H.L.; Peng, L.P. Genetic diversity and population structure of endangered endemic Paeonia jishanensis in China and conservation implications. Biochem. Syst. Ecol. 2003, 66, 319–325. [Google Scholar] [CrossRef]
- Zhang, D.Q.; Zhou, N. Genetic diversity and population structure of the endangered conifer Taxus wallichiana var. mairei (Taxaceae) revealed by Simple Sequence Repeat (SSR) markers. Biochem. Syst. Ecol. 2003, 49, 107–114. [Google Scholar]
- Zhang, Z.Y.; Wang, H.; Chen, W.; Pang, X.M.; Li, Y.Y. Genetic diversity and structure of native and non-native populations of the endangered plant Pinus dabeshanensis. Genet. Mol. Res. 2003, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Ohtani, M.; Suyama, Y.; Inomata, N.; Tsumura, Y.; Middleton, B.A.; Tachida, H.; Kusumi, J. Population genetic structure of a widespread coniferous tree, Taxodium distichum, [L.] Rich. (Cupressaceae), in the Mississippi River Alluvial Valley and Florida. Tree Genet. Gen. 2012, 8, 1135–1147. [Google Scholar] [CrossRef]
- Teixeira, H.; Nabais, C. Genetic diversity and differentiation of Juniperus thurifera in Spain and Morocco as determined by SSR. PLoS ONE 2014, 9, e88996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrick, J.L.; Godt, M. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 351, 1291–1298. [Google Scholar] [CrossRef]
- Schaal, B.A.; Hayworth, D.A.; Olsen, K.M.; Rauscher, J.T.; Smith, W.A. Phylogeographic studies in plants: Problems and prospects. Mol. Ecol. 1998, 7, 465–474. [Google Scholar] [CrossRef]
- Forest, F.; Grenyer, R.; Rouget, M.; Davies, T.J.; Cowling, R.M.; Faith, D.P.; Balmford, A.; Manning, J.C.; Proches, S.; van der Bank, M.; et al. Preserving the evolutionary potential of floras in biodiversity hotspots. Nature 1998, 445, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Berg, C.V.D.; Povoa, O.; Monteiro, A. Low genetic diversity and significant structuring in the endangered Mentha cervina populations and its implications for conservation. Biochem. Syst. Ecol. 2013, 50, 51–61. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, X.L.; Huang, Y.S. Preliminary report on provenance test of Thuja koraiensis Nakai. Shaanxi For. Sci. Technol. 2015, 3, 29–32. [Google Scholar]
- Heywood, V.H.; Iriondo, J.M. Plant conservation: Old problems, new perspectives. Biol. Conserv. 2015, 113, 321–335. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Populations | Location | Number | Longitude (E) | Latitude (N) | Altitude/m |
|---|---|---|---|---|---|
| LGZ | Lenggouzi, Changbai, Jilin | 155 | 126°28′ | 41°37′ | 1108 |
| SDG | Sandaogou, Baishan, Jilin | 26 | 126°28′ | 41°51′ | 483 |
| DJG | Dajinggou, Baishan, Jilin | 51 | 126°44′ | 41°52′ | 1172 |
| Feature | Value |
|---|---|
| Number of contigs | 875,792 |
| No. of contigs (≥200 bp) | 805,284 |
| No. of contigs (≥500 bp) | 2227 |
| No. of contigs (≥1000 bp) | 95 |
| N50 (bp) | 274 |
| GC (%) | 41.38 |
| Total contig length (bp) | 230,095,060 |
| Max. contig length (bp) | 4265 |
| Min. contig length (bp) | 150 |
| Average contig length (bp) | 262 |
| Searching Item | Number |
|---|---|
| Sequences examined | 875,792 |
| Size of examined sequences (bp) | 230,095,060 |
| Identified SSRs | 37,761 |
| SSR-containing sequences | 30,102 |
| Sequences containing more than one SSR | 5212 |
| SSRs present in compound formation | 7253 |
| Mononucleotide SSRs | 16,802 |
| Dinucleotide SSRs | 14,795 |
| Trinucleotide SSRs | 5119 |
| Tetranucleotide SSRs | 478 |
| Pentanucleotide SSRs | 133 |
| Hexanucleotide SSRs | 434 |
| SSR Name | Contigs | Primer Sequence (5′–>3′) | Motif | Tm (°C) | Size (bp) | NA | HO | HE | PIC | HWE |
|---|---|---|---|---|---|---|---|---|---|---|
| BFTK-39 | contig_120265 | F: TGTTCACTCCTCATCCACCG R: ACCGACATGATCTGCACACA | (TG)11 | 60 | 200 | 4 | 1.000 | 0.653 | 0.599 | ns |
| BFTK-51 | contig_169311 | F: TCATTGGAGTTGTATGGTGTCA R: TGCACAATTTGACCACTTGGA | (CT)34 | 59 | 159 | 3 | 0.571 | 0.561 | 0.465 | ns |
| BFTK-68 | contig_220530 | F: ACAACAAAGCGGTGGTAAACC R: GAATTGATGCTCAGCAGCCG | (GA)14 | 60 | 198 | 3 | 0.375 | 0.461 | 0.398 | ns |
| BFTK-123 | contig_387595 | F: TGCTTGCACTTGGATGTTGTG R: GCTCGATGCCAGGGTTTTTC | (TG)19 | 60 | 175 | 3 | 0.800 | 0.540 | 0.466 | ns |
| BFTK-136 | contig_412632 | F: CCCCCGGGCATAGATCAAAT R: CCCCCGGGCATAGATCAAAT | (TA)10 | 60 | 183 | 3 | 1.000 | 0.594 | 0.511 | ns |
| BFTK-167 | contig_487894 | F: TGAAGTCCCCATCTACATGTCA R: CTCAAACCAACTCCGTTACCT | (TG)16 | 59 | 169 | 3 | 0.125 | 0.664 | 0.590 | ** |
| BFTK-187 | contig_551058 | F: AGGACACAGAACAGAGCAGC R: CGGGTTAGCACATCAGGGAT | (AAC)10 | 60 | 147 | 2 | 0.750 | 0.469 | 0.359 | ns |
| BFTK-194 | contig_576296 | F: TACCTCGGAGATCAACCCCA R: CTCCCTCACATGGATGCCAA | (GA)13 | 60 | 109 | 2 | 0.143 | 0.133 | 0.124 | ns |
| BFTK-199 | contig_589312 | F: ATAGGGCACGACTAGCTTGC R: CATTCTTCAGCCTCCTGGTGT | (AC)15 | 60 | 132 | 4 | 0.667 | 0.667 | 0.620 | ns |
| BFTK-261 | contig_680582 | F: AGAGGTGGGGAAGAGGAGAC R: AGGCCCTAAACCCTATAACCA | (AG)9 | 60 | 142 | 5 | 0.286 | 0.653 | 0.602 | * |
| BFTK-273 | contig_689976 | F: TCCCATGTTTGTGGTCTCAGT R: TCCCCCAGAGTGCAACATTC | (TTG)9 | 60 | 209 | 7 | 1.000 | 0.820 | 0.798 | ns |
| BFTK-295 | contig_111296 | F: CGCAAGTCCAAATCAGCAAC R: TCGTGCAAACTTCCGTACCA | (CAA)6 | 59 | 146 | 2 | 0.750 | 0.500 | 0.375 | ns |
| Mean | 3.417 | 0.622 | 0.559 | 0.492 |
| Populations | NA | NE | I | HO | HE | F | PIC |
|---|---|---|---|---|---|---|---|
| LGZ | 7.667 | 2.557 | 1.091 | 0.766 | 0.572 | −0.279 | 0.503 |
| SDG | 3.667 | 2.750 | 1.024 | 0.629 | 0.582 | −0.068 | 0.504 |
| DJG | 5.111 | 2.235 | 0.908 | 0.537 | 0.491 | −0.036 | 0.428 |
| Mean | 5.481 | 2.514 | 1.008 | 0.644 | 0.548 | −0.128 | 0.478 |
| Source | df | SS | MS | Percentage of Variation (%) |
|---|---|---|---|---|
| Among Populations | 2 | 164.500 | 82.250 | 17% |
| Within Populations | 229 | 1489.435 | 6.504 | 83% |
| Total | 231 | 1653.935 | 88.754 | 100% |
| Populations | LGZ | SDG | DJG |
|---|---|---|---|
| LGZ | |||
| SDG | 0.061 | ||
| DJG | 0.048 | 0.078 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, L.; Cui, Y.; Li, X.; Chen, W.; Zhang, Z.; Pang, X.; Li, Y. Genetic Evaluation of Natural Populations of the Endangered Conifer Thuja koraiensis Using Microsatellite Markers by Restriction-Associated DNA Sequencing. Genes 2018, 9, 218. https://doi.org/10.3390/genes9040218
Hou L, Cui Y, Li X, Chen W, Zhang Z, Pang X, Li Y. Genetic Evaluation of Natural Populations of the Endangered Conifer Thuja koraiensis Using Microsatellite Markers by Restriction-Associated DNA Sequencing. Genes. 2018; 9(4):218. https://doi.org/10.3390/genes9040218
Chicago/Turabian StyleHou, Lu, Yanhong Cui, Xiang Li, Wu Chen, Zhiyong Zhang, Xiaoming Pang, and Yingyue Li. 2018. "Genetic Evaluation of Natural Populations of the Endangered Conifer Thuja koraiensis Using Microsatellite Markers by Restriction-Associated DNA Sequencing" Genes 9, no. 4: 218. https://doi.org/10.3390/genes9040218
APA StyleHou, L., Cui, Y., Li, X., Chen, W., Zhang, Z., Pang, X., & Li, Y. (2018). Genetic Evaluation of Natural Populations of the Endangered Conifer Thuja koraiensis Using Microsatellite Markers by Restriction-Associated DNA Sequencing. Genes, 9(4), 218. https://doi.org/10.3390/genes9040218