
Novel Selection Approaches to Identify Antibodies Targeting Neoepitopes on the C5b6 Intermediate Complex to Inhibit Membrane Attack Complex Formation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
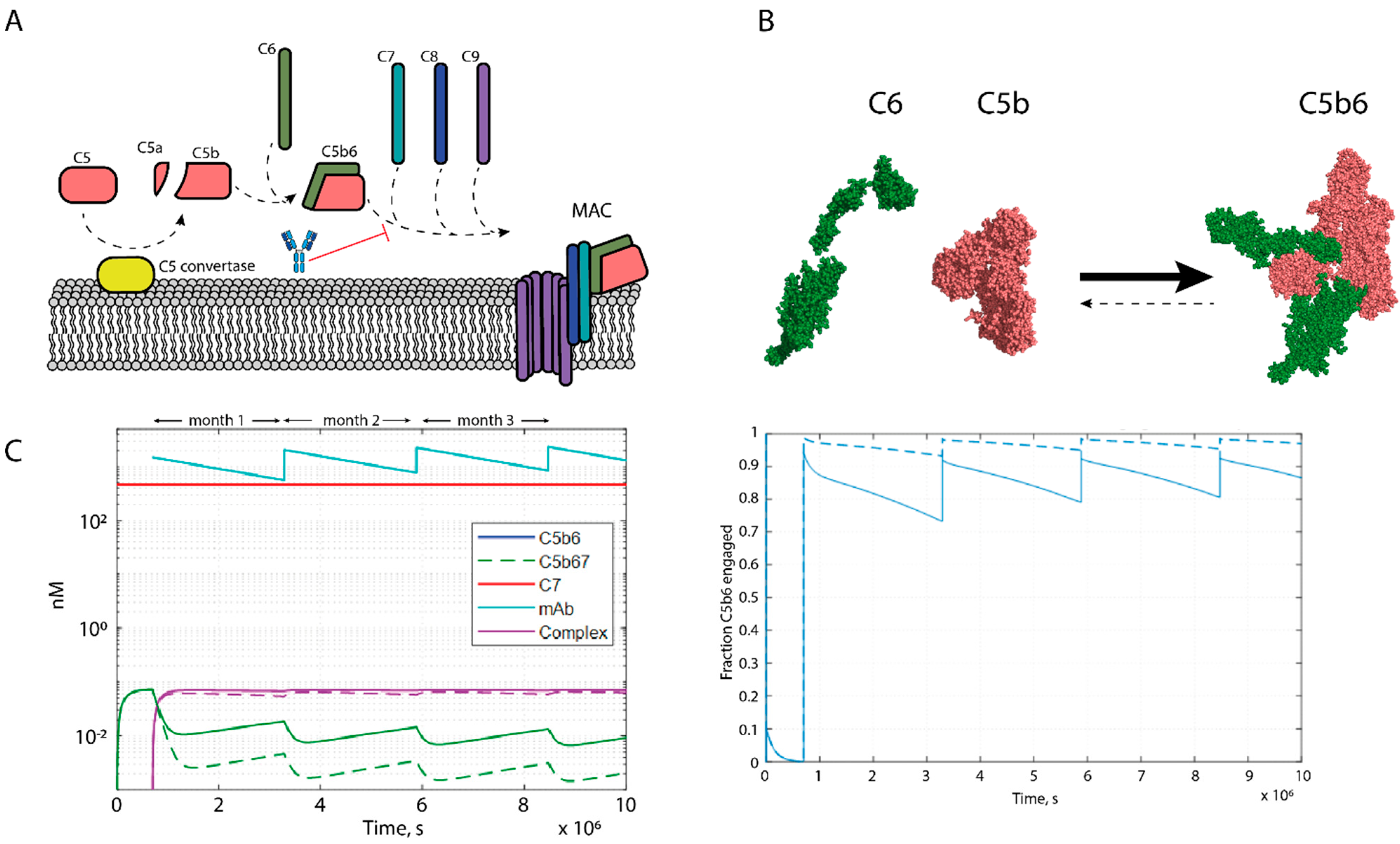
2.1. Complement System Dose Modelling
2.2. Antibody Selections
2.3. Antibody Expression and Purification
2.4. Cloning, Expression and Purification of an Anti-C5 and Anti-MAC Tool Antibodies
2.5. Biolayer Interferometry
2.6. Terminal Complement Assay
2.7. Surface Plasmon Resonance
2.8. C9 Oligomerization HTRF Assay
2.9. Liposome Leakage Assay
3. Results
3.1. Dose Prediction Modelling
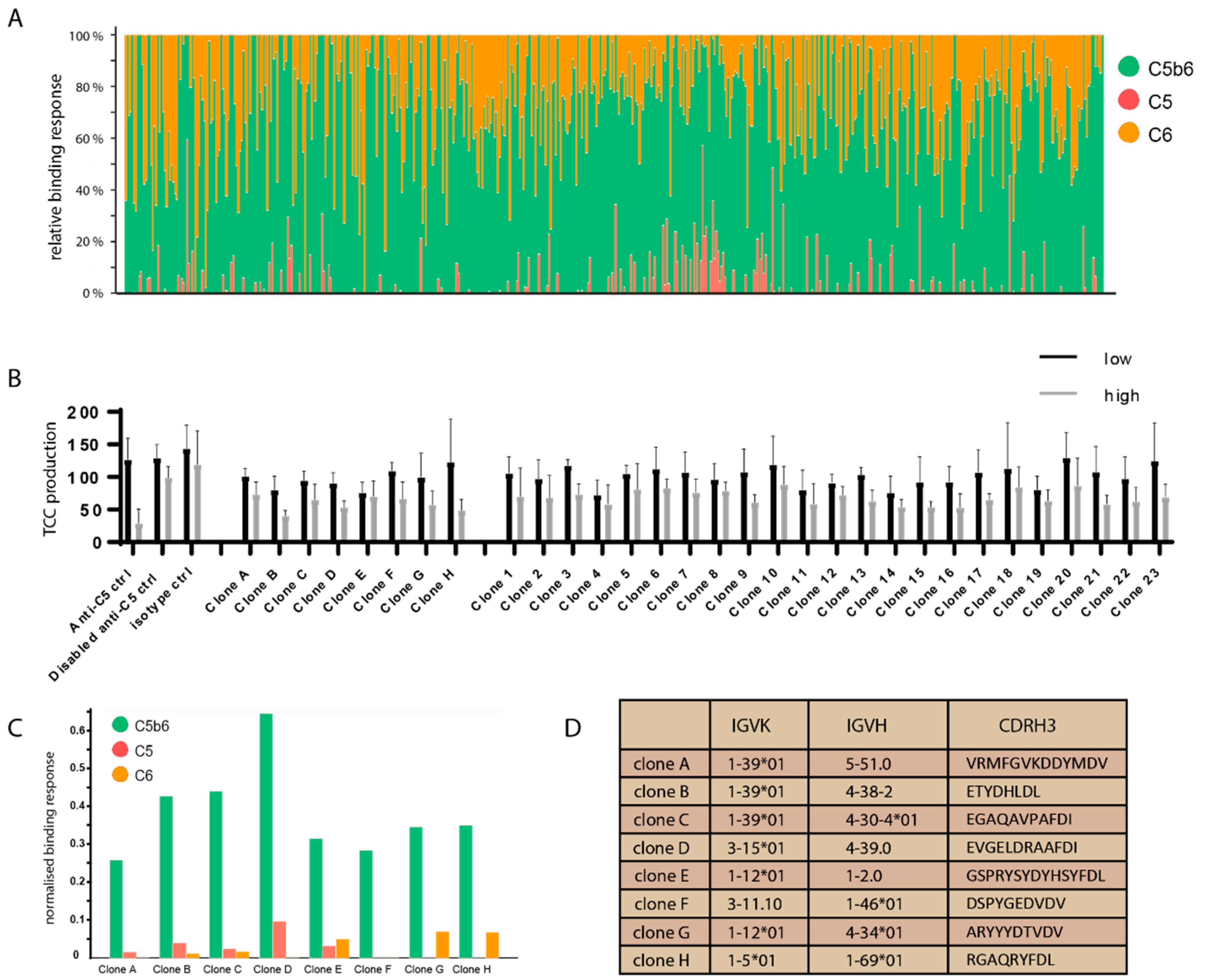
3.2. Naïve Antibody Library Selections
3.3. Characterisation of Naïve Antibody Library Selection Outputs
3.4. Affinity Maturation of Antibody Hit Panel
3.5. Development of Antigen Binding and Complement Function Assays to Screen Affinity Matured Antibodies
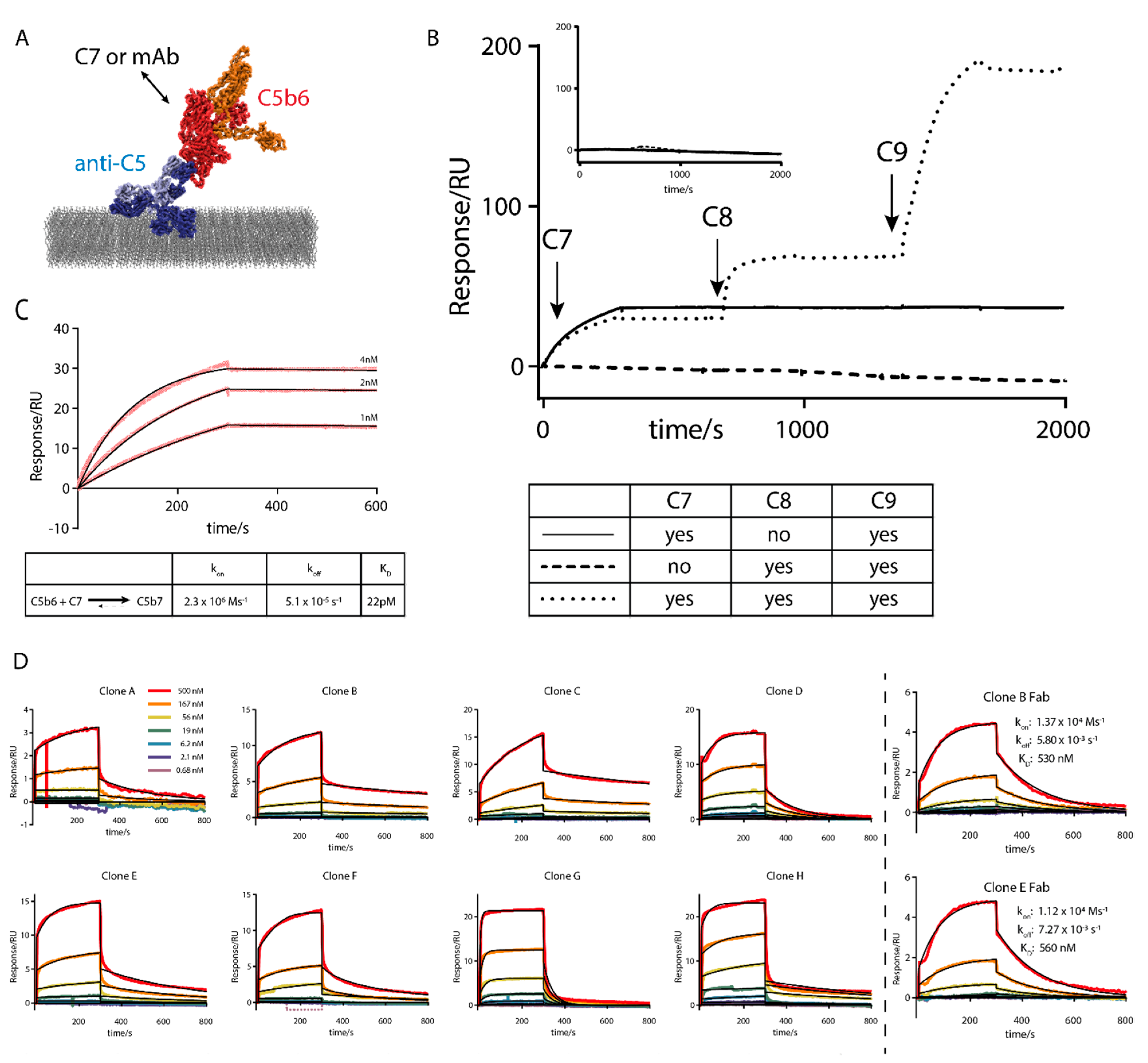
3.5.1. Development of a Surface Plasmon Resonance Assay Format Suitable for C5b6
3.5.2. Development of Functional Complement Assays
3.6. Characterisation of the Affinity Matured Antibody Output and Comparison with Parental Antibodies
3.6.1. Antigen Binding Properties of Affinity Matured Antibodies
3.6.2. Functional Characterisation of Affinity Matured Antibodies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M.J. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000, 192, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.K.S.; Kavanagh, D. Diseases of complement dysregulation-an overview. Semin. Immunopathol. 2018, 40, 49–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, B.P. The role of complement in neurological and neuropsychiatric diseases. Expert Rev. Clin. Immunol. 2015, 11, 1109–1119. [Google Scholar] [CrossRef]
- Harris, C.L. Expanding horizons in complement drug discovery: Challenges and emerging strategies. Semin. Immunopathol. 2018, 40, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, T.C.; Rollins, S.A.; Rother, R.P.; Giannoni, M.A.; Hartman, S.L.; Elliott, E.A.; Nye, S.H.; Matis, L.A.; Squinto, S.P.; Evans, M.J. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996, 33, 1389–1401. [Google Scholar] [CrossRef]
- Hillmen, P.; Hall, C.; Marsh, J.C.; Elebute, M.; Bombara, M.P.; Petro, B.E.; Cullen, M.J.; Richards, S.J.; Rollins, S.A.; Mojcik, C.F.; et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2004, 350, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, D.; Yu, Z.X.; Zhang, Y.; Patel, R.; Sun, F.; Lasaro, M.A.; Bouchard, K.; Andrien, B.; Marozsan, A.; Wang, Y.; et al. Design and preclinical characterization of ALXN1210: A novel anti-C5 antibody with extended duration of action. PLoS ONE 2018, 13, e0195909. [Google Scholar] [CrossRef] [Green Version]
- Mannes, M.; Dopler, A.; Zolk, O.; Lang, S.J.; Halbgebauer, R.; Hochsmann, B.; Skerra, A.; Braun, C.K.; Huber-Lang, M.; Schrezenmeier, H.; et al. Complement inhibition at the level of C3 or C5: Mechanistic reasons for ongoing terminal pathway activity. Blood 2021, 137, 443–455. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Pawluczkowycz, A.W.; Peng, W.; Kennedy, A.D.; Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Selective and efficient inhibition of the alternative pathway of complement by a mAb that recognizes C3b/iC3b. Mol. Immunol. 2006, 43, 1010–1019. [Google Scholar] [CrossRef]
- Paixao-Cavalcante, D.; Torreira, E.; Lindorfer, M.A.; Rodriguez de Cordoba, S.; Morgan, B.P.; Taylor, R.P.; Llorca, O.; Harris, C.L. A humanized antibody that regulates the alternative pathway convertase: Potential for therapy of renal disease associated with nephritic factors. J. Immunol. 2014, 192, 4844–4851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katschke, K.J., Jr.; Stawicki, S.; Yin, J.; Steffek, M.; Xi, H.; Sturgeon, L.; Hass, P.E.; Loyet, K.M.; Deforge, L.; Wu, Y.; et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 2009, 284, 10473–10479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedemann, N.C.; Habel, M.; Ziereisen, J.; Hermann, M.; Schneider, C.; Wehling, C.; Kirschfink, M.; Kentouche, K.; Guo, R. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin. Immunol. 2017, 180, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhao, G.; Liu, C.; Fan, W.; Zhou, X.; Zeng, L.; Guo, Y.; Kou, Z.; Yu, H.; Li, J.; et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin. Infect. Dis. 2015, 60, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Morgan, B.P.; Daniels, R.H.; Watts, M.J.; Williams, B.D. In vivo and in vitro evidence of cell recovery from complement attack in rheumatoid synovium. Clin. Exp. Immunol. 1988, 73, 467–472. [Google Scholar]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126 Pt 13, 2903–2913. [Google Scholar] [CrossRef] [Green Version]
- Podack, E.R.; Kolb, W.P.; Muller-Eberhard, H.J. The C5b-6 complex: Formation, isolation, and inhibition of its activity by lipoprotein and the S-protein of human serum. J. Immunol. 1978, 120, 1841–1848. [Google Scholar] [PubMed]
- Podack, E.R. Molecular composition of the tubular structure of the membrane attack complex of complement. J. Biol. Chem. 1984, 259, 8641–8647. [Google Scholar] [CrossRef]
- Korotaevskiy, A.A.; Hanin, L.G.; Khanin, M.A. Non-linear dynamics of the complement system activation. Math. Biosci. 2009, 222, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Thai, C.T.; Ogata, R.T. Recombinant C345C and factor I modules of complement components C5 and C7 inhibit C7 incorporation into the complement membrane attack complex. J. Immunol. 2005, 174, 6227–6232. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Roach, W.; Sun, T.; Jain, T.; Prinz, B.; Yu, T.Y.; Torrey, J.; Thomas, J.; Bobrowicz, P.; Vasquez, M.; et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: A FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 2013, 26, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef]
- Jore, M.M.; Johnson, S.; Sheppard, D.; Barber, N.M.; Li, Y.I.; Nunn, M.A.; Elmlund, H.; Lea, S.M. Structural basis for therapeutic inhibition of complement C5. Nat. Struct. Mol. Biol. 2016, 23, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Schatz-Jakobsen, J.A.; Zhang, Y.; Johnson, K.; Neill, A.; Sheridan, D.; Andersen, G.R. Structural Basis for Eculizumab-Mediated Inhibition of the Complement Terminal Pathway. J. Immunol. 2016, 197, 337–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faudry, E.; Perdu, C.; Attree, I. Pore formation by T3SS translocators: Liposome leakage assay. Methods Mol. Biol. 2013, 966, 173–185. [Google Scholar]
- Serna, M.; Giles, J.L.; Morgan, B.P.; Bubeck, D. Structural basis of complement membrane attack complex formation. Nat. Commun. 2016, 7, 10587. [Google Scholar] [CrossRef] [Green Version]
- Hansch, G.M.; Hammer, C.H.; Vanguri, P.; Shin, M.L. Homologous species restriction in lysis of erythrocytes by terminal complement proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 5118–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, K.M.; Arndt, K.M.; Pluckthun, A. Model and simulation of multivalent binding to fixed ligands. Anal. Biochem. 1998, 261, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosic, L.; Sutherland, W.M.; Kurek, J.; Edberg, J.C.; Taylor, R.P. Preparation of monoclonal antibodies to C3b by immunization with C3b(i)-sepharose. J. Immunol. Methods 1989, 120, 241–249. [Google Scholar] [CrossRef]
- Zelek, W.M.; Morgan, B. Monoclonal Antibodies Capable of Inhibiting Complement Downstream of C5 in Multiple Species. Front. Immunol. 2020, 11, 612402. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stach, L.; Dinley, E.K.H.; Tournier, N.; Bingham, R.P.; Gormley, D.A.; Bramhall, J.L.; Taylor, A.; Clarkson, J.E.; Welbeck, K.A.; Harris, C.L.; et al. Novel Selection Approaches to Identify Antibodies Targeting Neoepitopes on the C5b6 Intermediate Complex to Inhibit Membrane Attack Complex Formation. Antibodies 2021, 10, 39. https://doi.org/10.3390/antib10040039
Stach L, Dinley EKH, Tournier N, Bingham RP, Gormley DA, Bramhall JL, Taylor A, Clarkson JE, Welbeck KA, Harris CL, et al. Novel Selection Approaches to Identify Antibodies Targeting Neoepitopes on the C5b6 Intermediate Complex to Inhibit Membrane Attack Complex Formation. Antibodies. 2021; 10(4):39. https://doi.org/10.3390/antib10040039
Chicago/Turabian StyleStach, Lasse, Emily K. H. Dinley, Nadia Tournier, Ryan P. Bingham, Darren A. Gormley, Jo L. Bramhall, Adam Taylor, Jane E. Clarkson, Katherine A. Welbeck, Claire L. Harris, and et al. 2021. "Novel Selection Approaches to Identify Antibodies Targeting Neoepitopes on the C5b6 Intermediate Complex to Inhibit Membrane Attack Complex Formation" Antibodies 10, no. 4: 39. https://doi.org/10.3390/antib10040039
APA StyleStach, L., Dinley, E. K. H., Tournier, N., Bingham, R. P., Gormley, D. A., Bramhall, J. L., Taylor, A., Clarkson, J. E., Welbeck, K. A., Harris, C. L., Feeney, M., Hughes, J. P., Sepp, A., Batuwangala, T. D., Kitchen, S. J., & Nichols, E. -M. (2021). Novel Selection Approaches to Identify Antibodies Targeting Neoepitopes on the C5b6 Intermediate Complex to Inhibit Membrane Attack Complex Formation. Antibodies, 10(4), 39. https://doi.org/10.3390/antib10040039