Mineralogy, Geochemistry and Genesis of Agate—A Review


Abstract
:1. Introduction
2. Materials and Methods
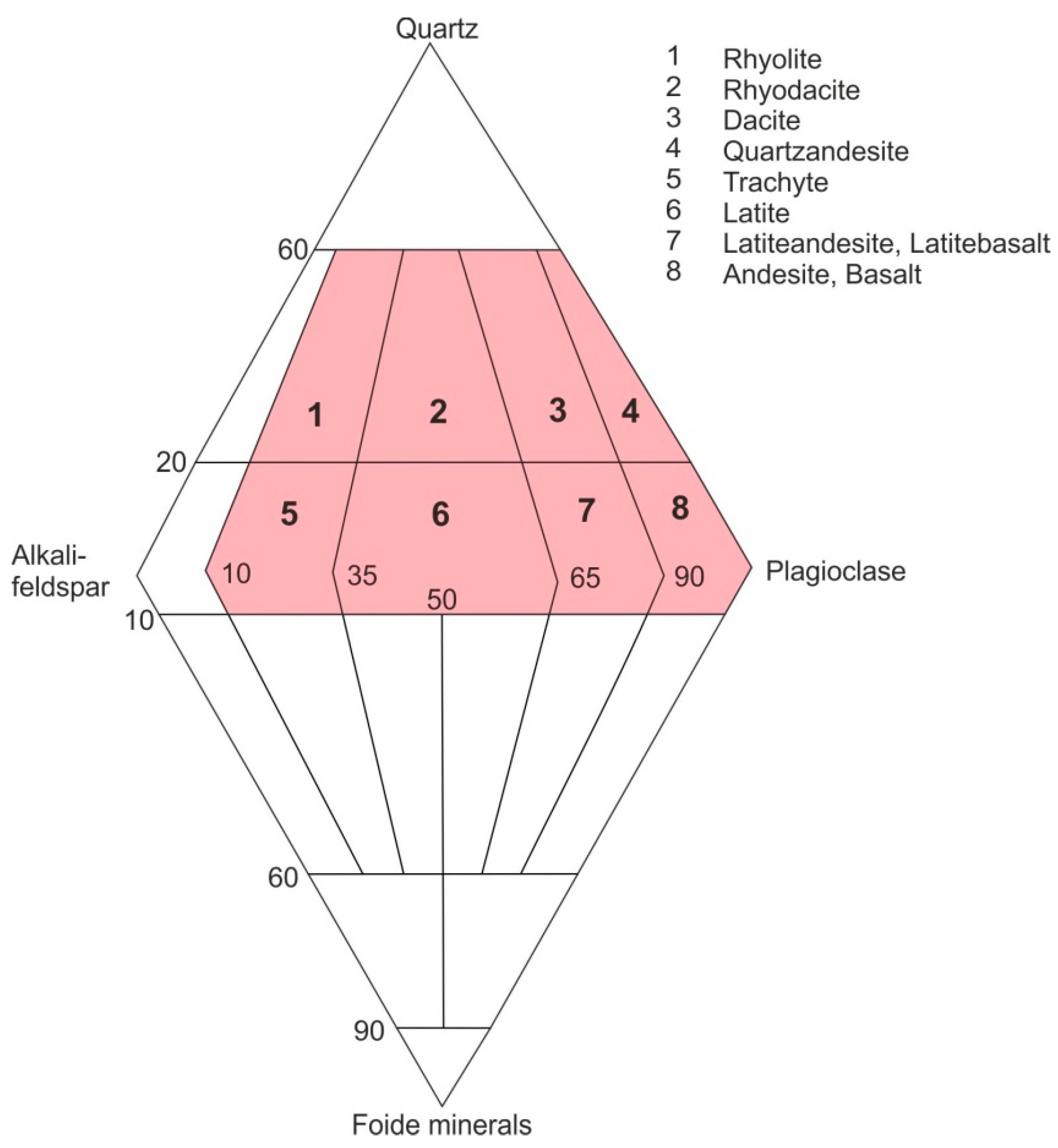
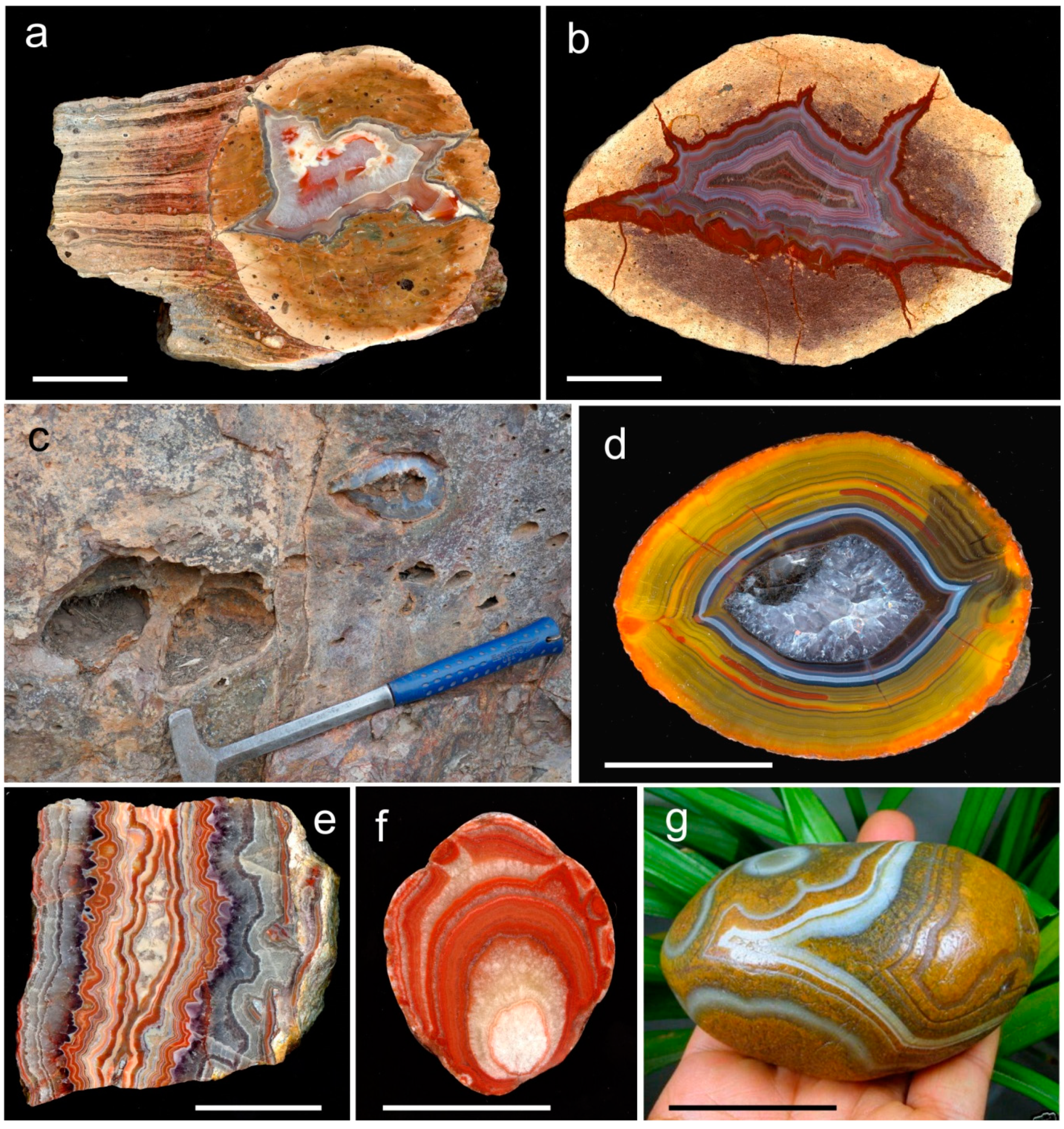
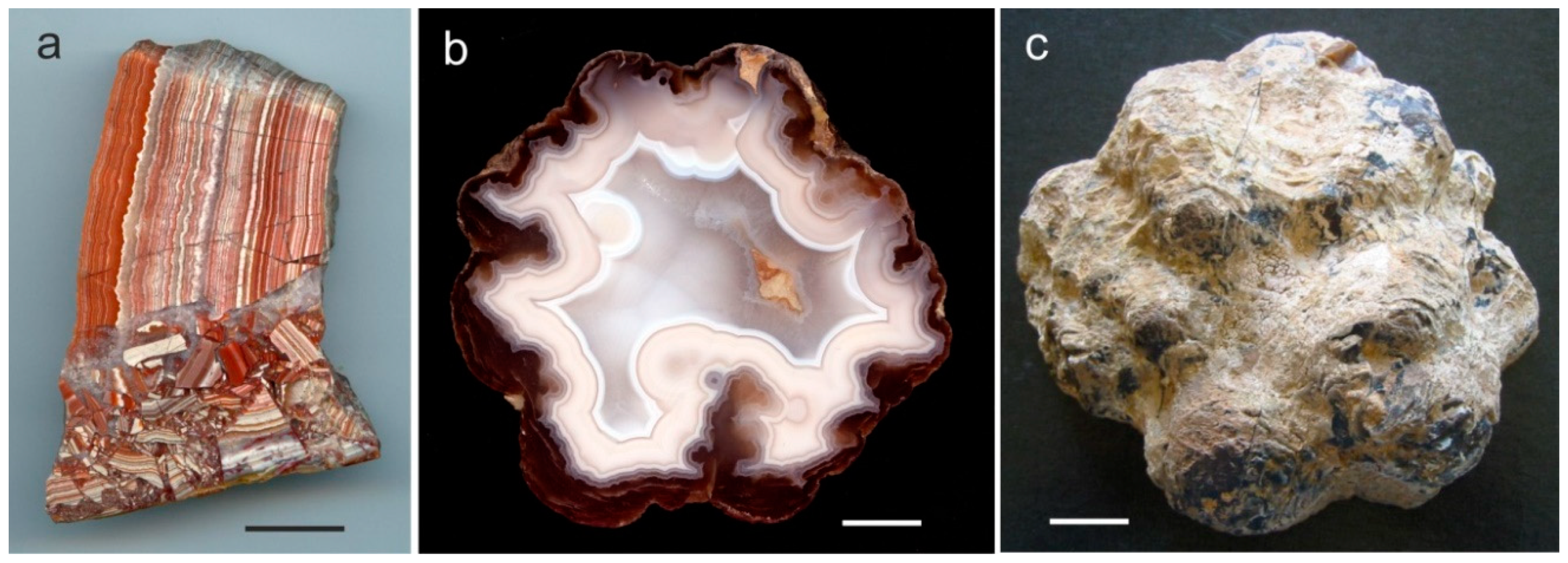
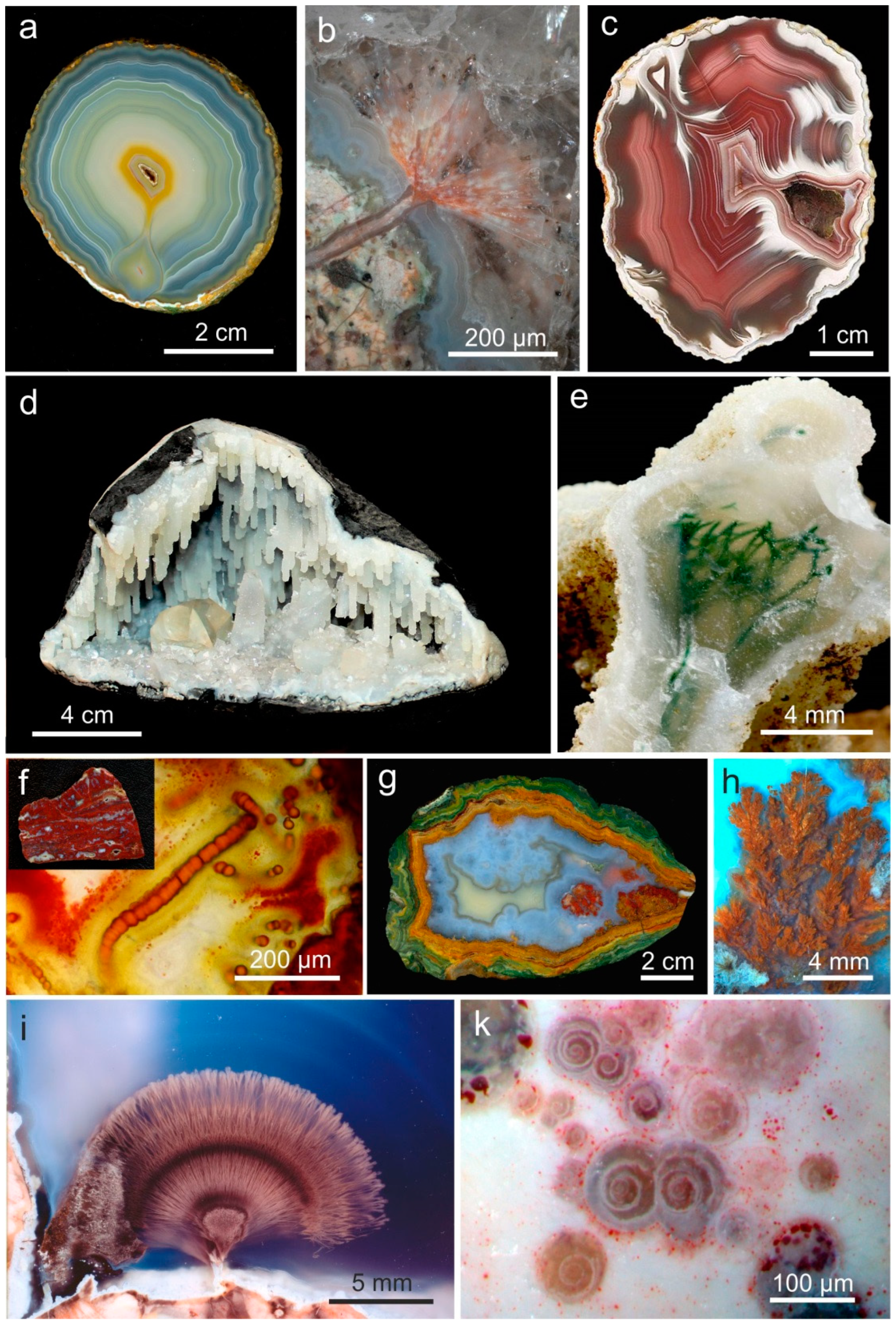
3. Geological Occurrences and Types of Agates
4. Agate Properties
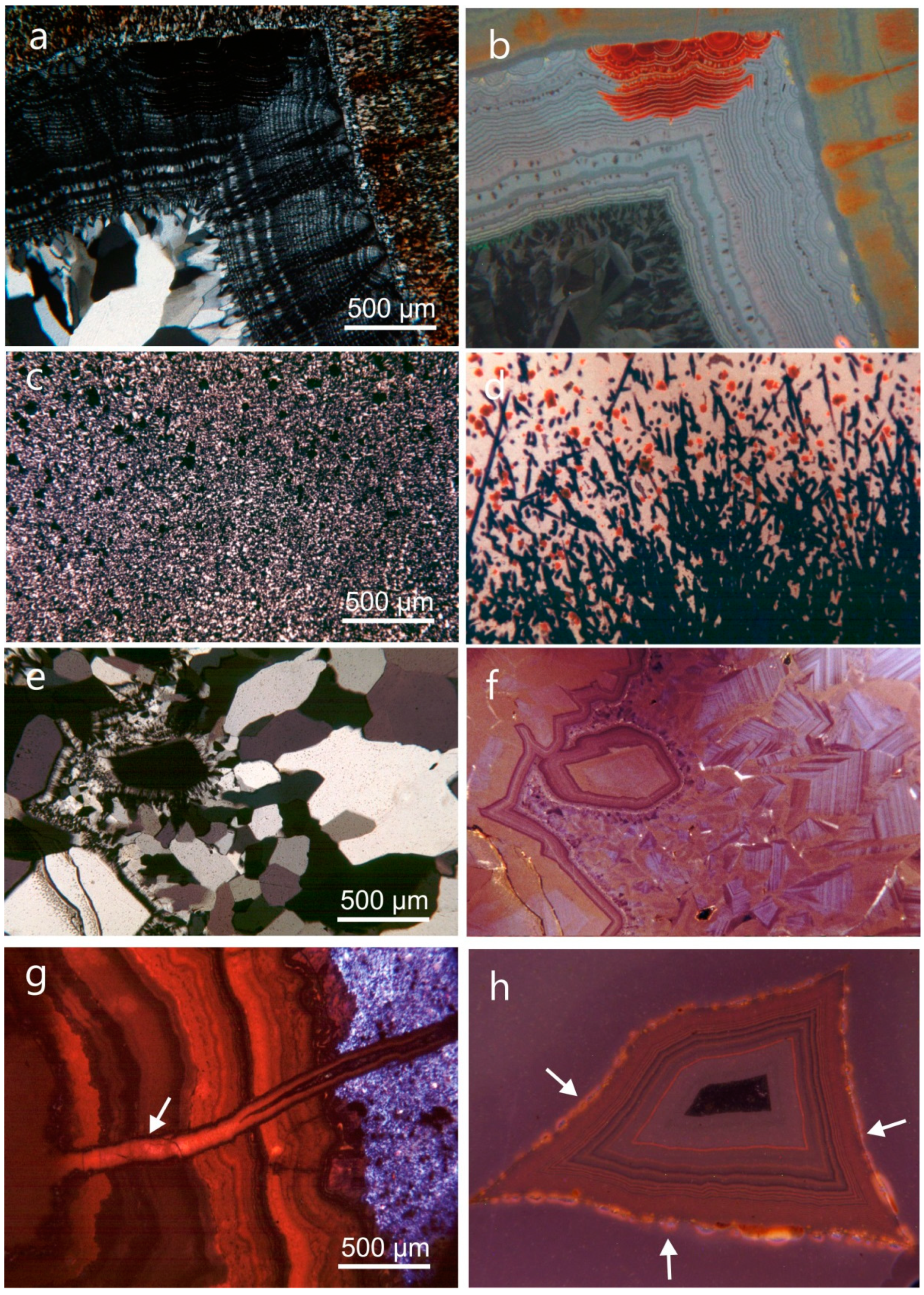
4.1. Mineralogy and Micro-Structure of Agates
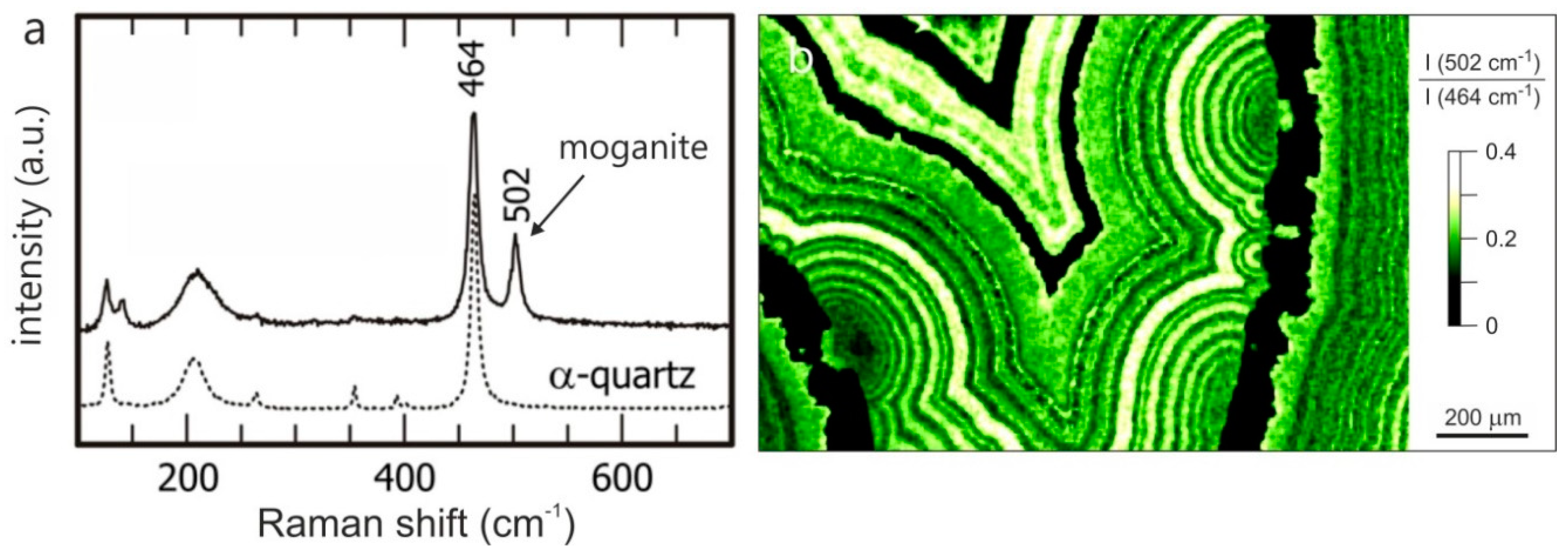
4.1.1. SiO2 Phases in Agate
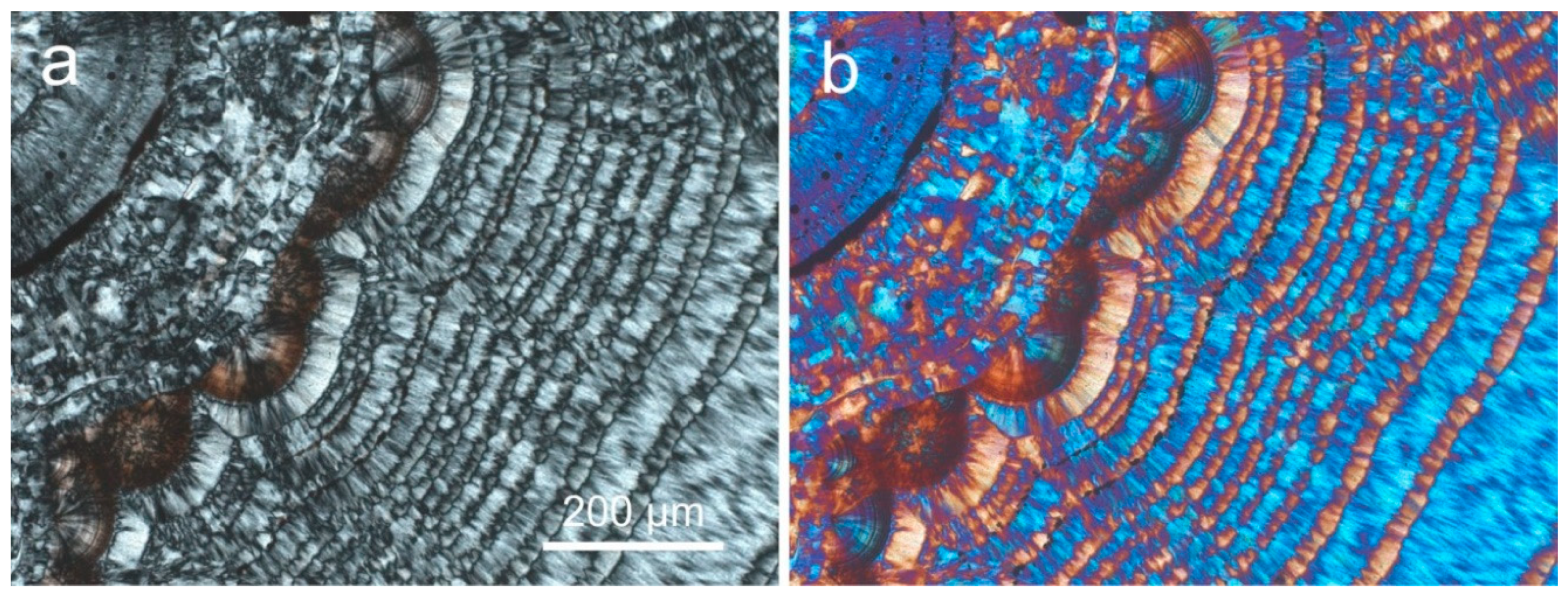
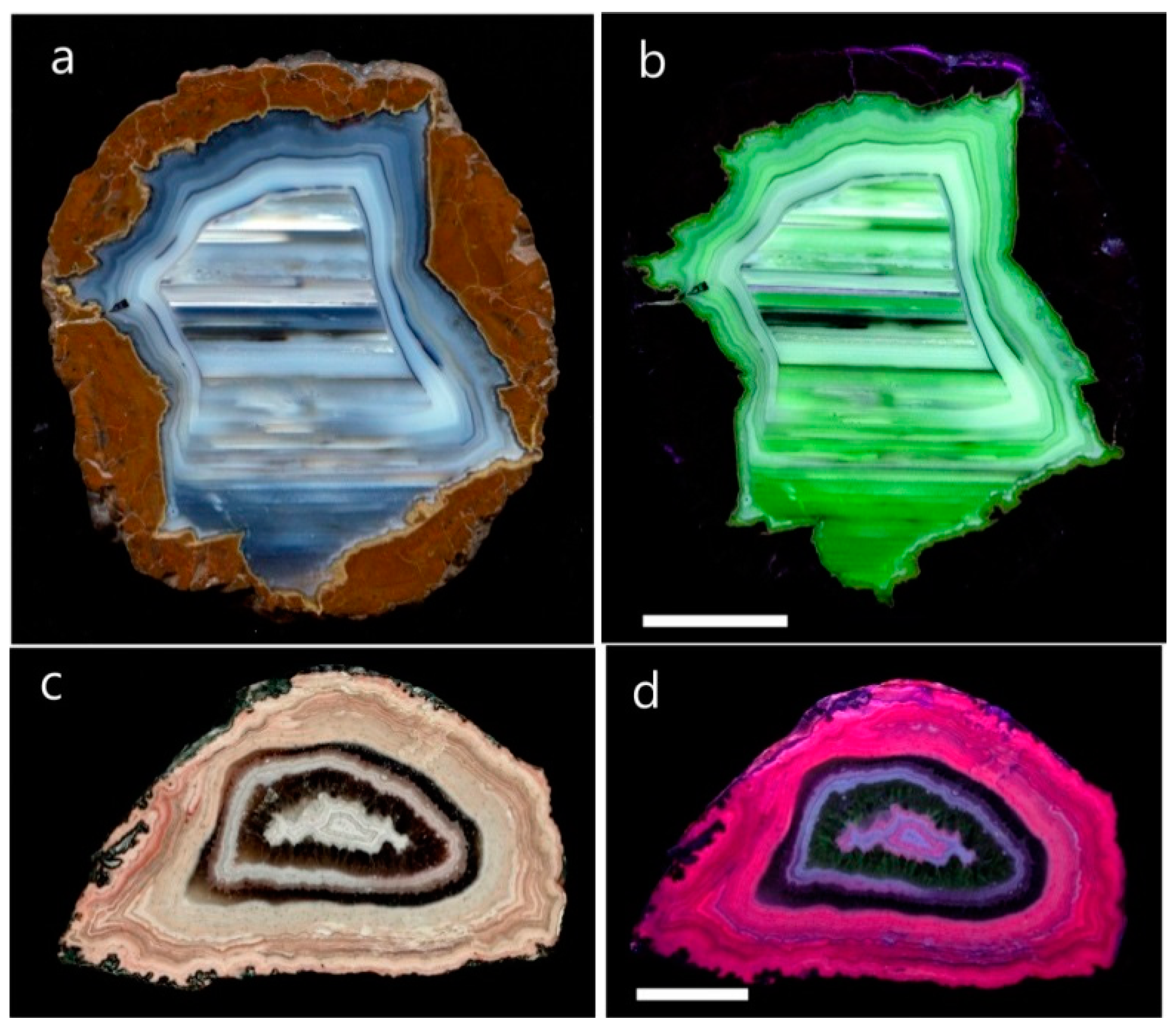
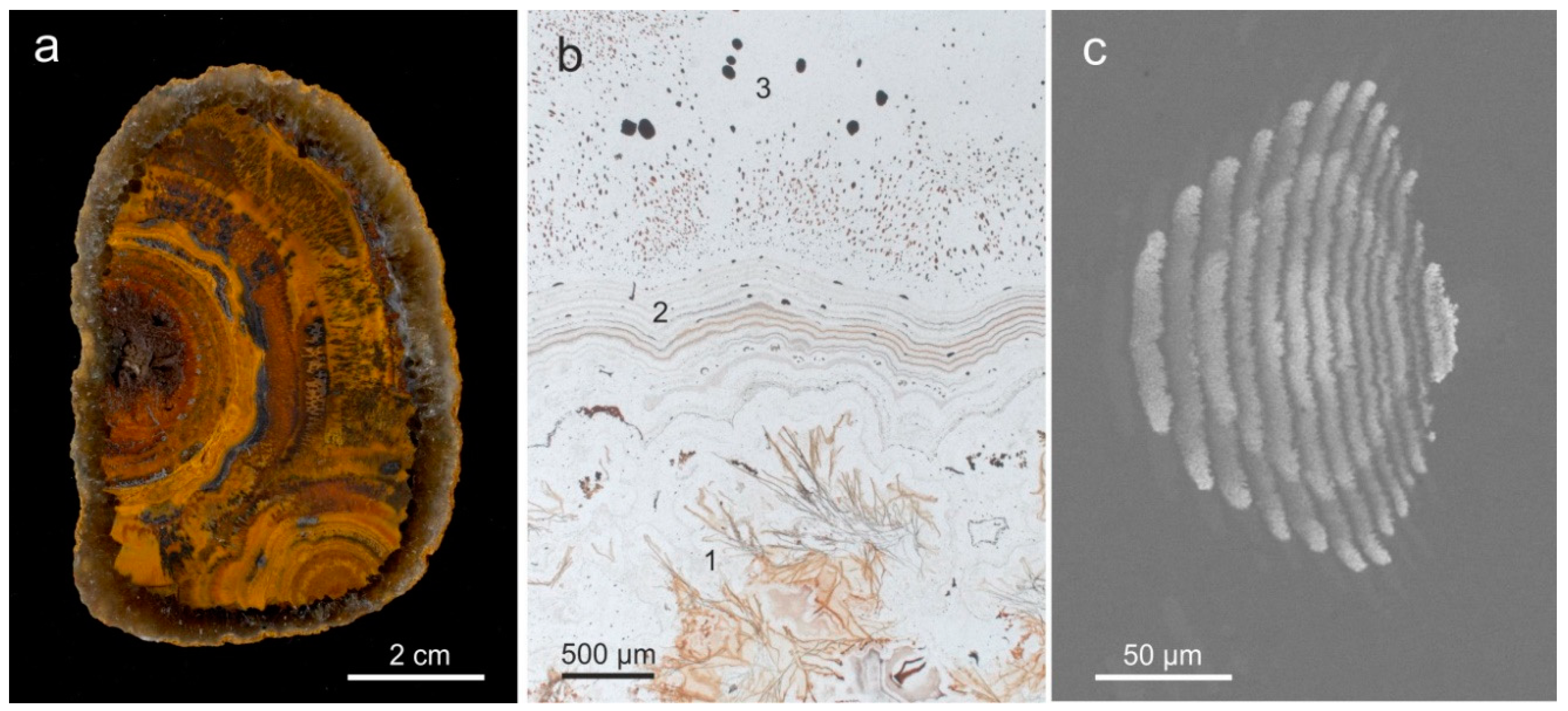
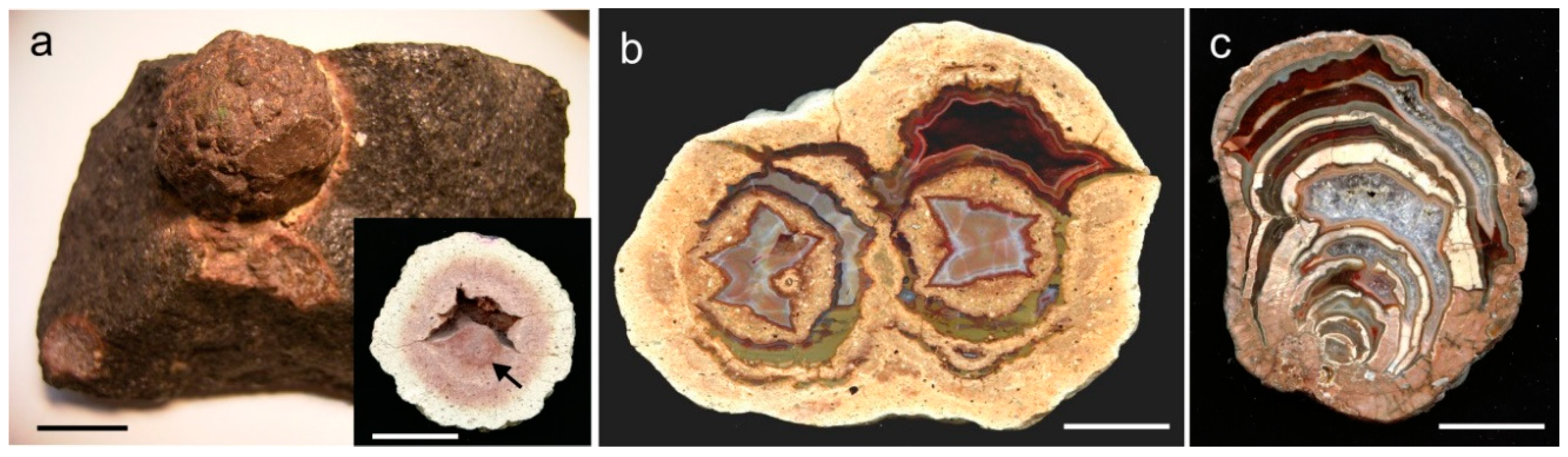
4.1.2. Microstructure of Agates and Agate Banding
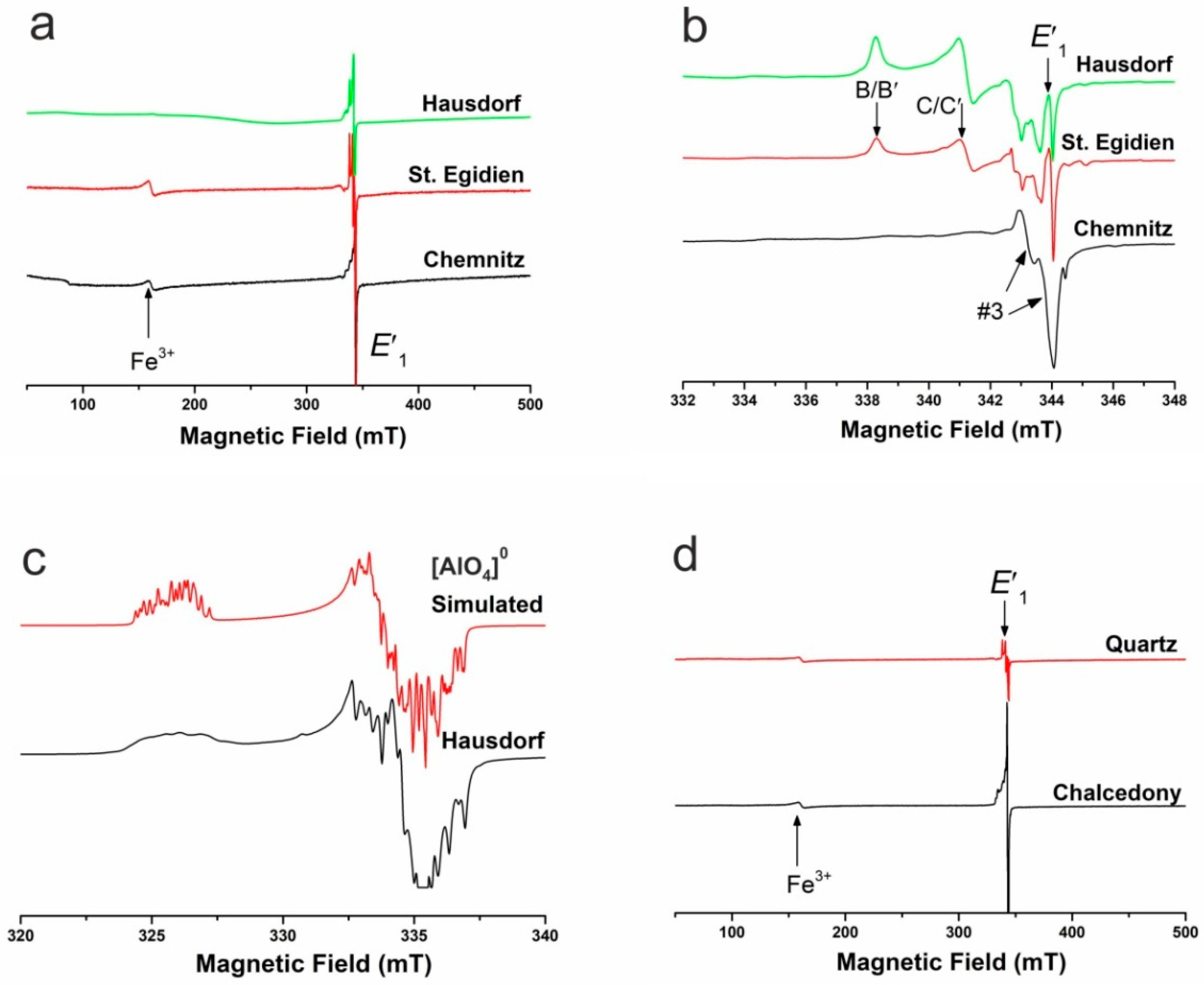
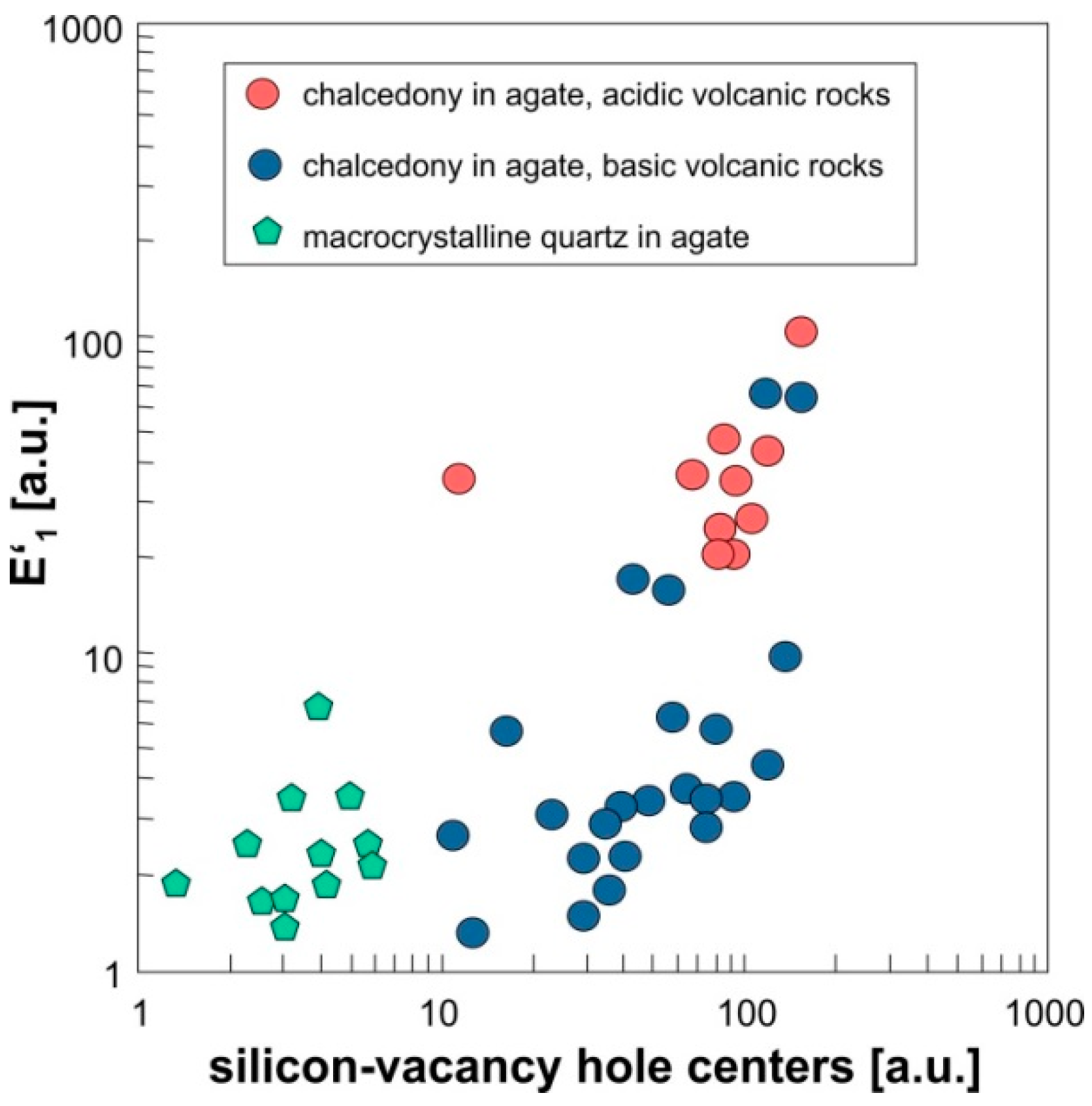
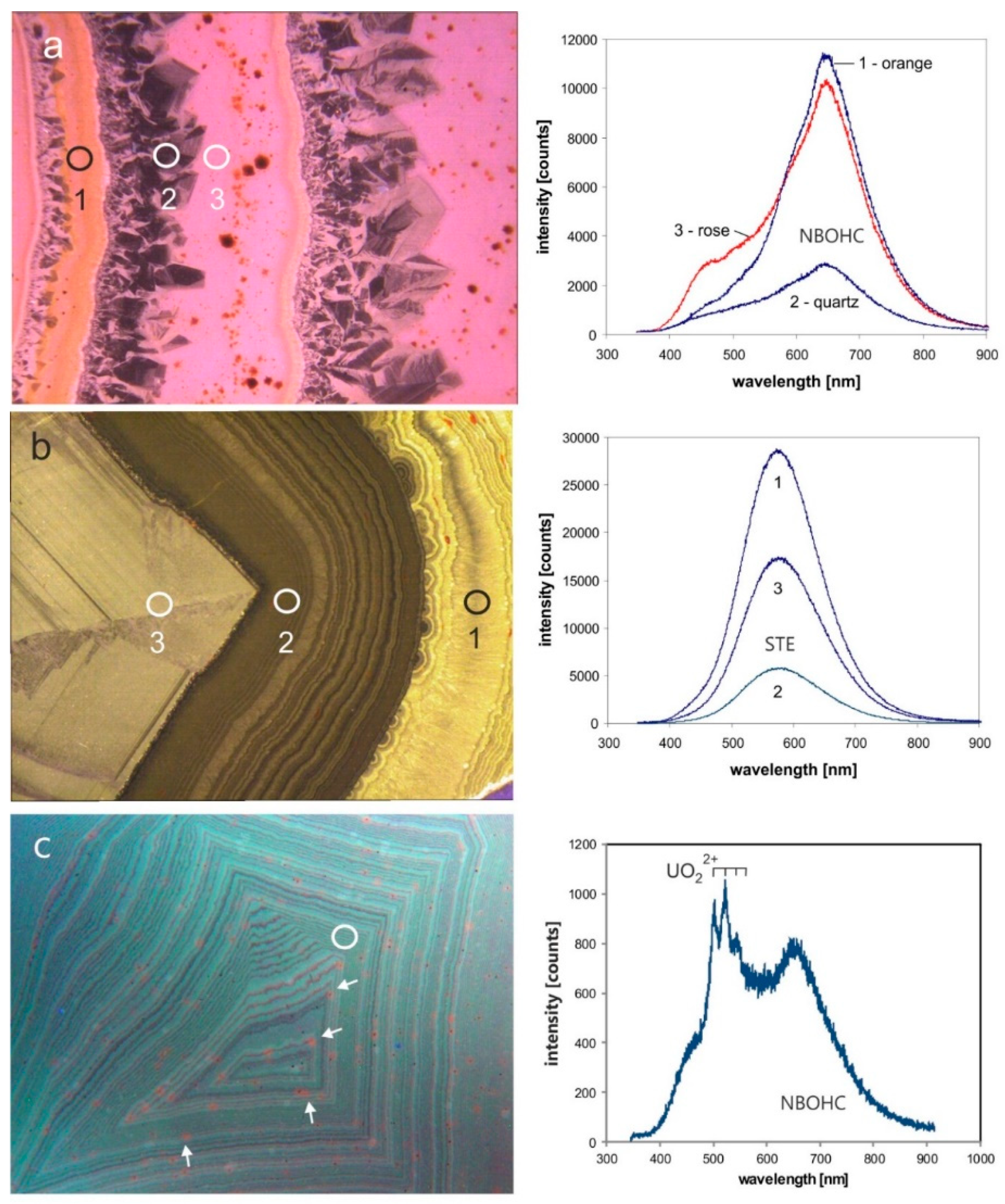
4.1.3. Point Defects in Agate
4.2. Geochemistry of Agates
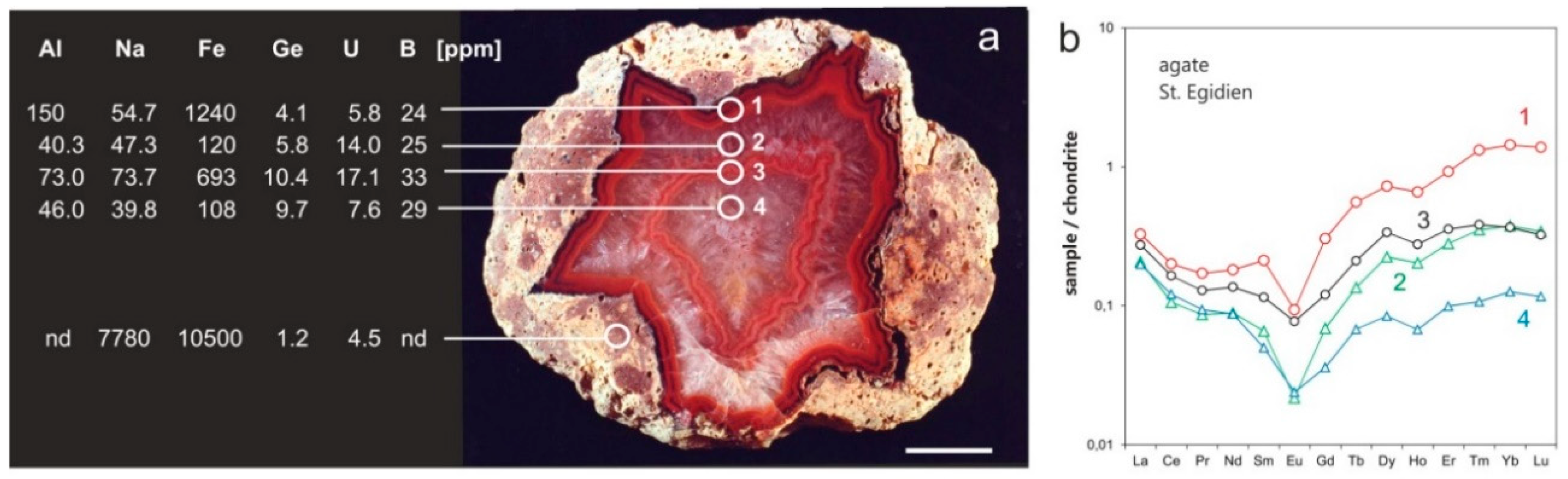
4.2.1. Trace Elements
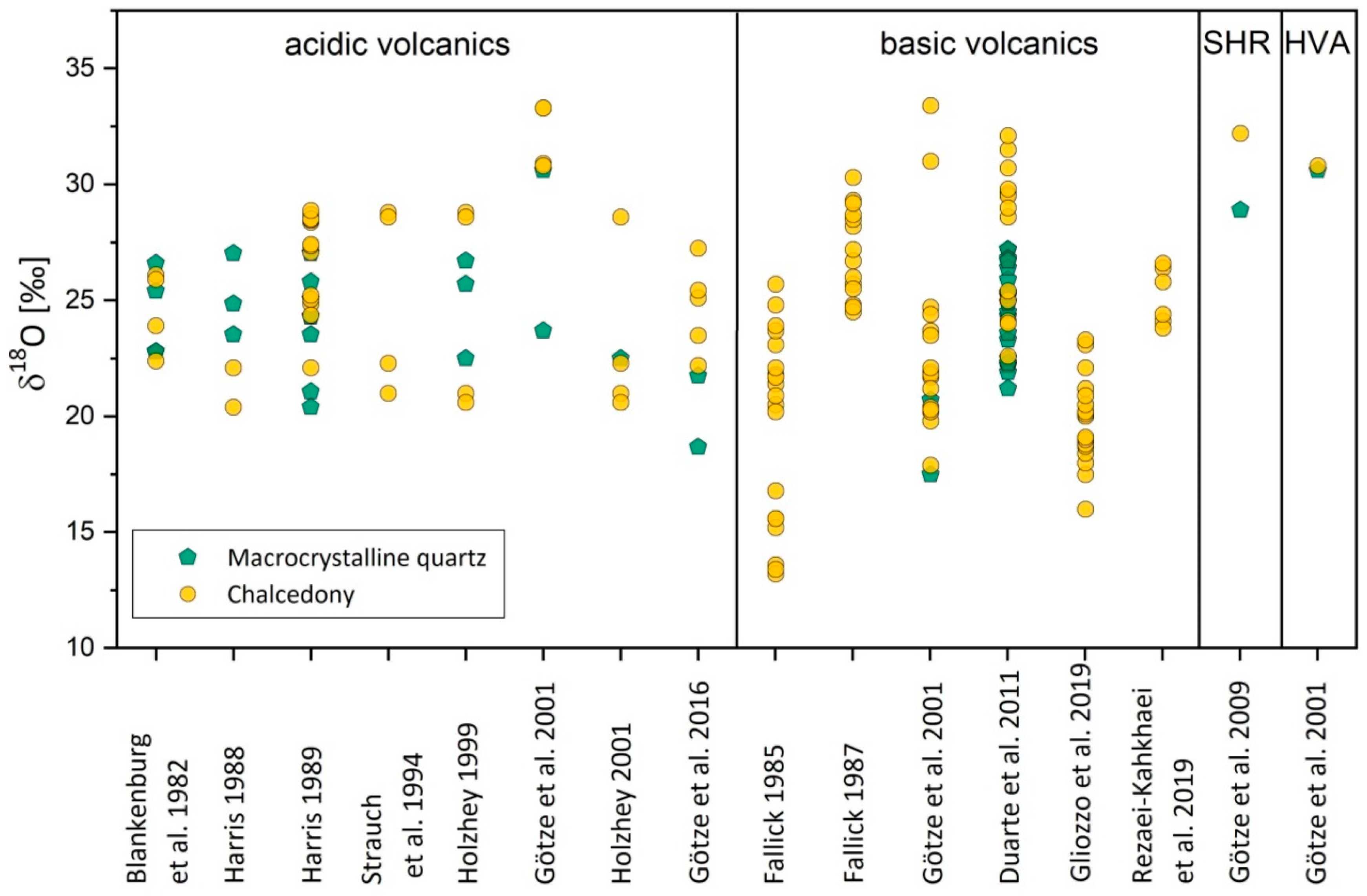
4.2.2. Isotope Composition
4.2.3. Fluid Inclusions and Water in Agate
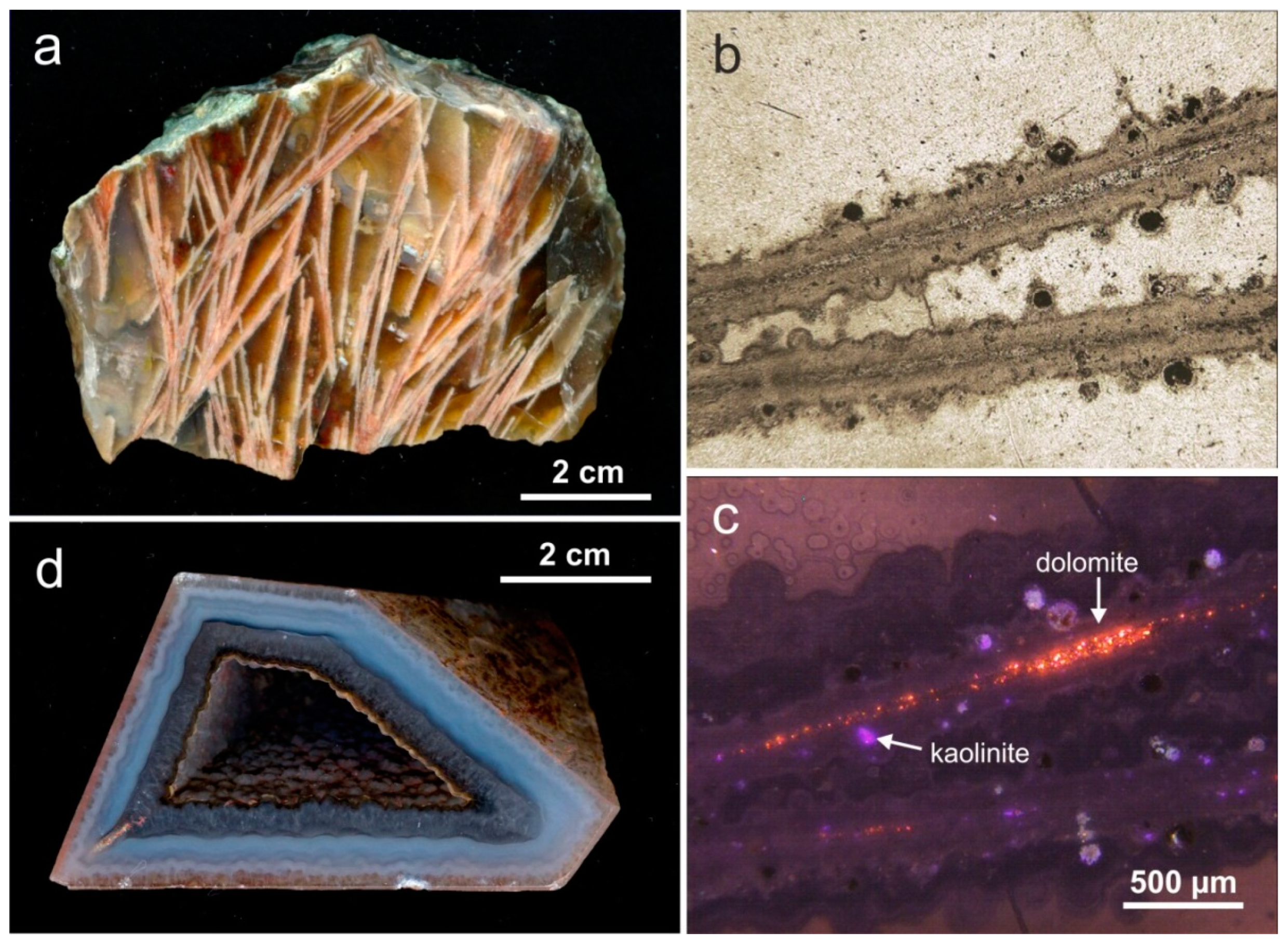
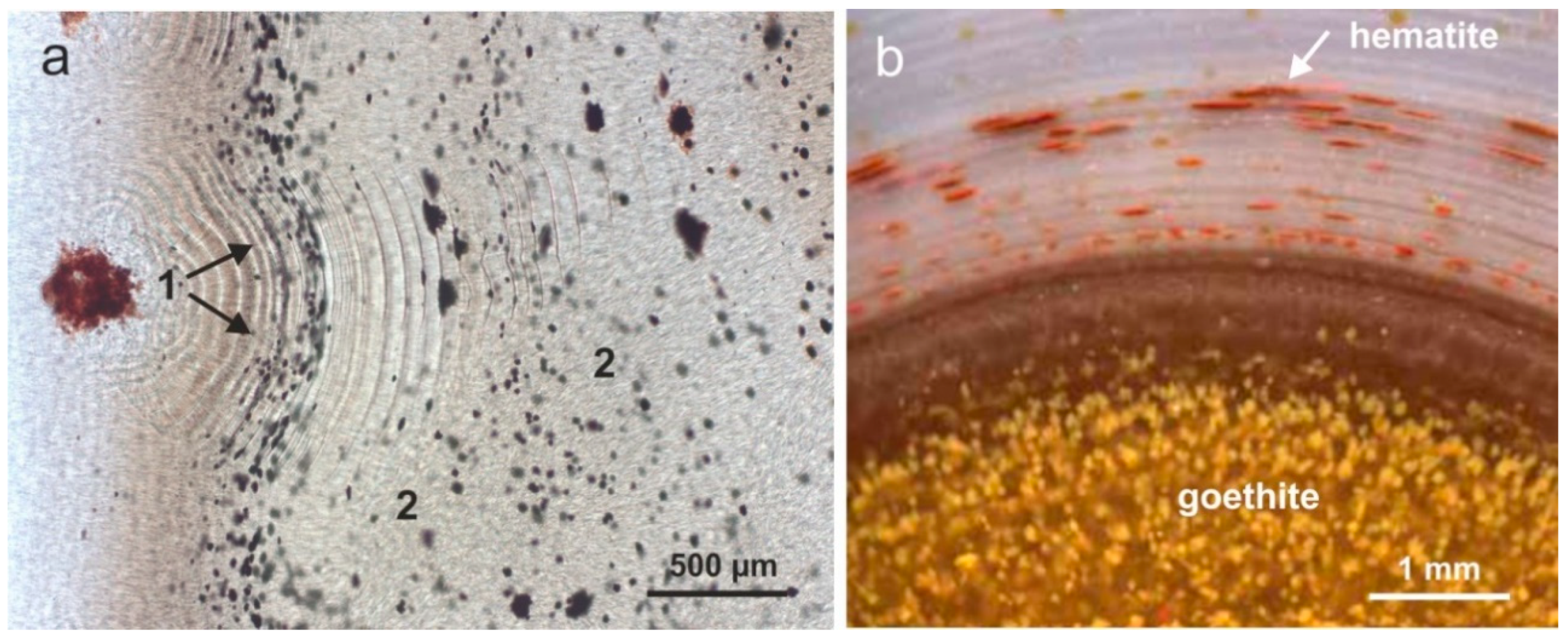
4.3. Paragenetic Minerals in Agates
5. Discussion
5.1. Origin of Cavities for Agate Formation
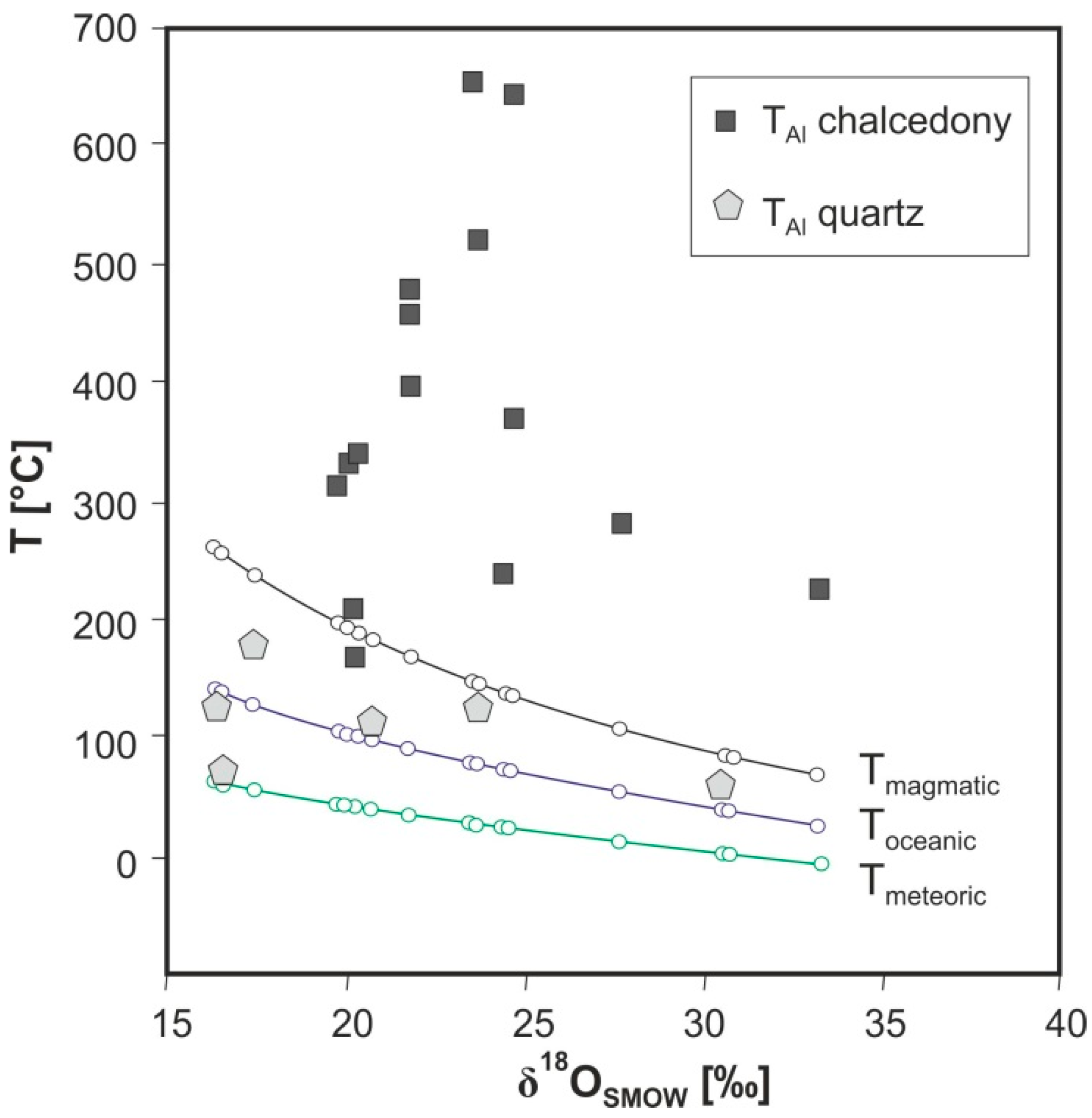
5.2. Temperature of Agate Formation
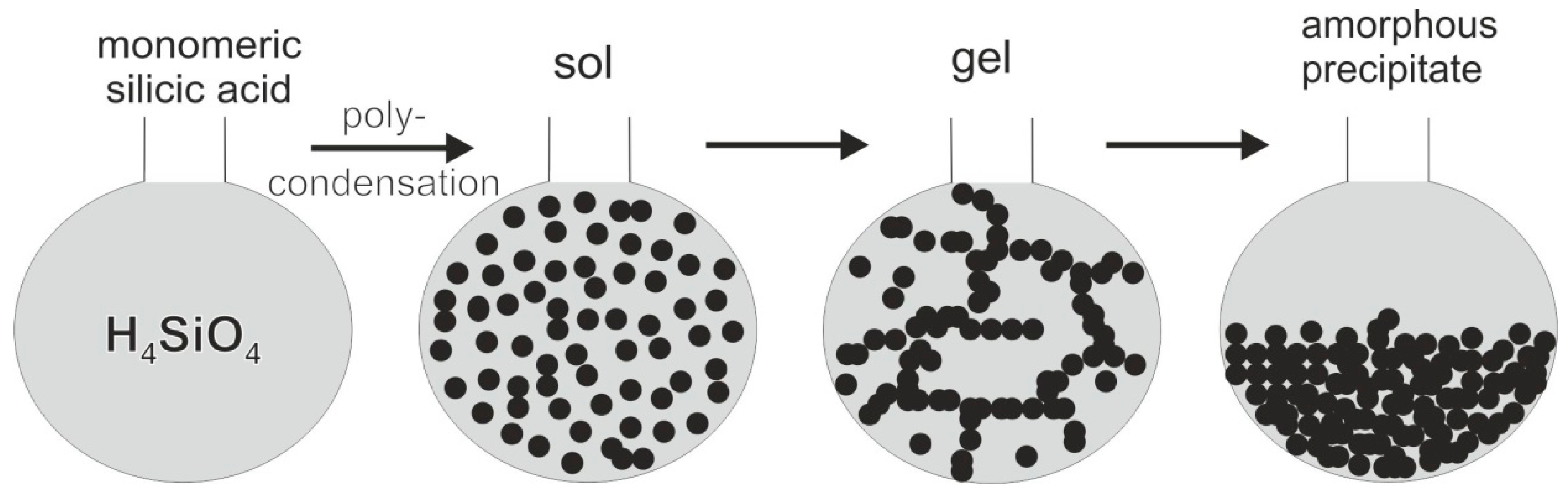
5.3. Origin and Supply of Silica
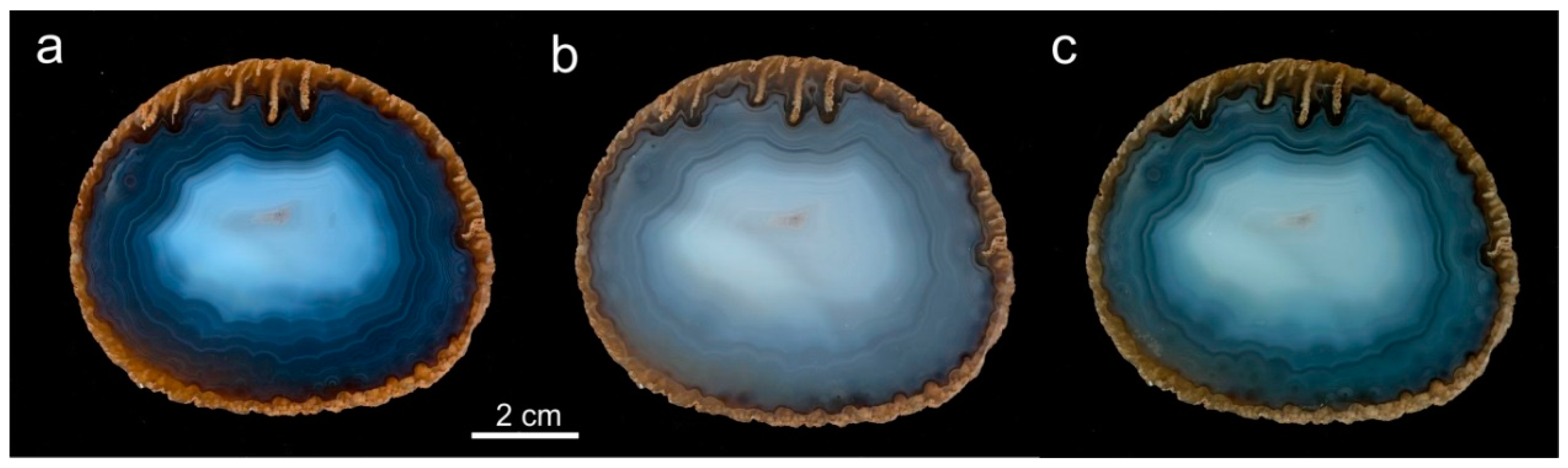
5.4. Formation of Agate Banding and Color
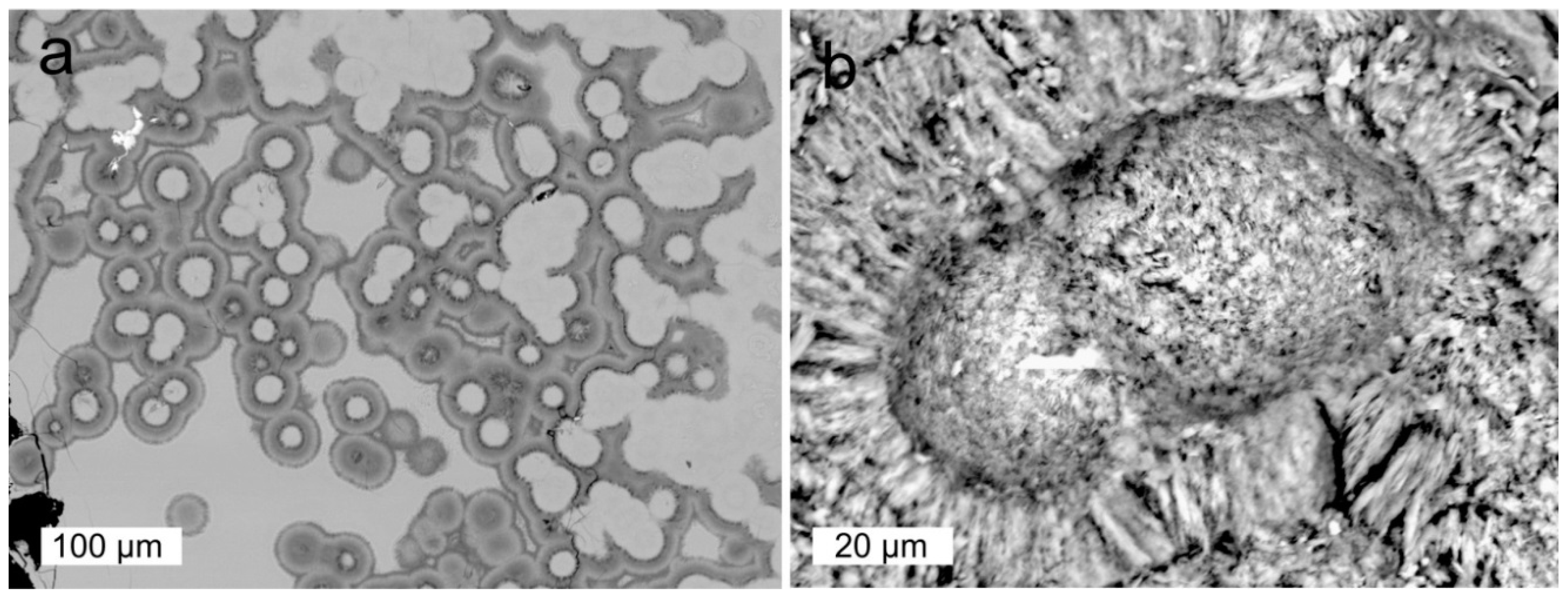
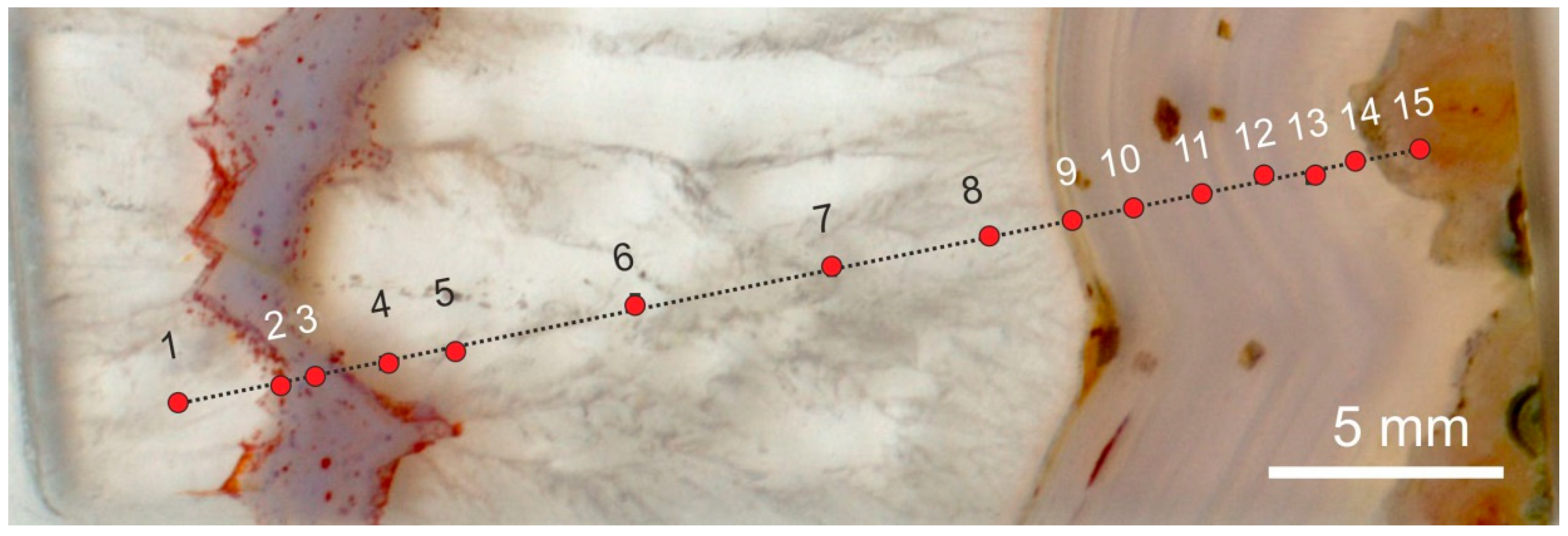
5.5. Microstructural Peculiarities and Possible Biosignatures in Agates
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blankenburg, H.-J. Achat; VEB Deutscher Verlag für Grundstoffindustrie: Leipzig, Germany, 1988; 203p. [Google Scholar]
- Zenz, J. Achate/Agate; Bode-Verlag: Haltern, Germany, 2005; 656p. [Google Scholar]
- Zenz, J. Achate/Agates II; Bode-Verlag: Haltern, Germany, 2009; 656p. [Google Scholar]
- Zenz, J. Achate/Agates III; Bode-Verlag: Haltern, Germany, 2011; 656p. [Google Scholar]
- Brückmann, U.F.B. Abhandlung von Edelsteinen; Waisenhaus-Buchhandlung: Braunschweig, Germany, 1773. [Google Scholar]
- Collini, C. Tagebuch Einer Reise, Welches Verschiedene Mineralogische Beobachtungen, Besonders Über Die Achate und Den. Basalt Enthält; C.F. Schwan: Mannheim, Germany, 1776; 582p. [Google Scholar]
- Noeggerath, M. On the porosity and colouring of agates, chalcedonies, etc. Edinb. New Philos. J. 1850, 58, 166–172. [Google Scholar]
- Landmesser, M. Das Problem der Achatgenese. Mitt. Pollichia 1984, 72, 5–137. [Google Scholar]
- Daubrée, A. Synthetische Studien zur Experimental-Geologie; Vieweg-Verlag: Braunschweig, Germany, 1880; 122p. [Google Scholar]
- Nacken, R. Über die Nachbildung von Chalzedon-Mandeln. Nat. und Volk. 1948, 78, 2–8. [Google Scholar]
- Liesegang, R.E. Die Entstehung der Achate. Zent. für Mineral. 1910, 11, 593–597. [Google Scholar]
- Liesegang, R.E. Die Achate; Verlag von Theodor Steinkopff: Dresden/Leipzig, Germany, 1915; 122p. [Google Scholar]
- Flörke, O.W.; Köhler-Herbertz, B.; Langer, K.; Tönges, I. Water in Microcrystalline Quartz of Volcanic Origin: Agates. Contrib. Mineral. Petrol. 1982, 80, 324–333. [Google Scholar] [CrossRef]
- Fallick, A.E.; Jocelyn, J.; Donnelly, T.; Guy, M.; Behan, C. Origin of agates in volcanic rocks from Scotland. Nature 1985, 313, 672–674. [Google Scholar] [CrossRef]
- Godovikov, A.A.; Ripinen, O.I.; Motorin, S.G. Agaty; Nedra: Moskva, Russia, 1987; 368p. [Google Scholar]
- Macpherson, H.-G. Agates; The Natural History Museum and the National Museums of Scotland: London, UK, 1989; 72p. [Google Scholar]
- Moxon, T. On the origin of agate with particular reference to fortification agate found in the Midland Valley, Scotland. Chem. der Erde 1991, 51, 251–260. [Google Scholar]
- Moxon, T. Agate: Microstructure and Possible Origin; Terra Publications: Doncaster, UK, 1996; 106p. [Google Scholar]
- Moxon, T. Agates: A study of ageing. Eur. J. Mineral. 2002, 14, 1109–1118. [Google Scholar] [CrossRef]
- Moxon, T. Studies on Agate—Microscopy, Spectroscopy, Growth, High Temperature and Possible Origin; Terra Publications: Doncaster, UK, 2009; 102p. [Google Scholar]
- Moxon, T. A re-examination of water in agate and its bearing on the agate genesis enigma. Mineral. Mag. 2017, 81, 1223–1244. [Google Scholar] [CrossRef]
- Holzhey, G. Vorkommen und Genese der Achate und Paragensemineralien in Rhyolithkugeln aus Rotliegendvulkaniten des Thüringer Waldes. Ph.D. Thesis, TU Bergakademie, Freiberg, Germany, 1993; 132p. [Google Scholar]
- Heany, P.J. A proposed mechanism for the growth of chalcedony. Contrib. Mineral. Petrol. 1993, 115, 66–74. [Google Scholar] [CrossRef]
- Pabian, R.K.; Zarins, A. Banded Agates—Origins and Inclusions; Educational Circular No. 12; University of Nebraska: Lincoln, RI, USA, 1994; 32p. [Google Scholar]
- Ortoleva, P.; Chen, Y.; Chen, W. Agates, Geodes, Concretions and Orbicules: Self-Organized Zoning and Morphology; Kruhl, J.H., Ed.; Springer: Heidelberg, Germany, 1994; pp. 283–305. [Google Scholar]
- Heaney, P.J.; Davis, A.M. Observation and origin of self-organized textures in agates. Science 1995, 269, 1562–1565. [Google Scholar] [CrossRef]
- Merino, E.; Wang, Y.; Deloule, E. Genesis of agates in flood basalts: Twisting of chalcedony fibers and trace-element geochemistry. Am. J. Sci. 1995, 295, 1156–1176. [Google Scholar] [CrossRef]
- Cross, B.L. The Agates of Northern Mexico; Burgess Publishing Division: Edina, MI, USA, 1996; 130p. [Google Scholar]
- Götze, J.; Nasdala, L.; Kleeberg, R.; Wenzel, M. Occurrence and distribution of “moganite” in agate/chalcedony: A combined micro-Raman, Rietveld, and cathodoluminescence study. Contrib. Mineral. Petrol. 1998, 133, 96–105. [Google Scholar] [CrossRef]
- Götze, J.; Tichomirowa, M.; Fuchs, H.; Pilot, J.; Sharp, Z. Geochemistry of agates: A trace element and stable isotope study. Chem. Geol. 2001, 175, 523–541. [Google Scholar] [CrossRef]
- Götze, J.; Möckel, R.; Kempe, U.; Kapitonov, I.; Vennemann, T. Origin and characteristics of agates in sedimentary rocks from the Dryhead area, Montana/USA. Mineral. Mag. 2009, 73, 673–690. [Google Scholar] [CrossRef]
- Götze, J.; Martins, M.S.; Czarnobay, J.C. Achate aus Brasilien. Veröff. Mus. für Nat. Chemnitz 2010, 33, 63–78. [Google Scholar]
- Götze, J.; Müller, A.; Polgári, M.; Pál-Molnár, E. Biosignaturen in Achat/Chalcedon–die Rolle von Mikroorganismen bei der Bildung von SiO2. Mineralienwelt 2011, 22, 90–96. [Google Scholar]
- Götze, J.; Schrön, W.; Möckel, R.; Heide, K. The role of fluids in the formation of agate. Geochemistry 2012, 72, 283–286. [Google Scholar] [CrossRef]
- Götze, J.; Nasdala, L.; Kempe, U.; Libowitzky, E.; Rericha, A.; Vennemann, T. Origin of black colouration in onyx agate from Mali. Mineral. Mag. 2012, 76, 115–127. [Google Scholar] [CrossRef]
- Götze, J.; Gaft, M.; Möckel, R. Uranium and uranyl luminescence in agate/chalcedony. Mineral. Mag. 2015, 79, 983–993. [Google Scholar] [CrossRef]
- Götze, J.; Möckel, R.; Vennemann, T.; Müller, A. Origin and geochemistry of agates from Permian volcanic rocks of the Sub-Erzgebirge basin (Saxony, Germany). Chem. Geol. 2016, 428, 77–91. [Google Scholar] [CrossRef]
- Götze, J.; Möckel, R.; Eulitz, B. “Karbonat-Achat” von Krásný Dvoreček. Mineralienwelt 2018, 4, 82–87. [Google Scholar]
- Götze, J.; Berek, H.; Schäfer, K. Micro-structural phenomena in agate/chalcedony: Spiral growth. Mineral. Mag. 2019, 83, 281–291. [Google Scholar] [CrossRef]
- Moxon, T.; Rios, S. Moganite and water content as a function of age in agate: An XRD and thermogravimetric study. Eur. J. Mineral. 2004, 4, 693–706. [Google Scholar] [CrossRef]
- Petránek, J. Entstehung von gravitations und adhäsionsgebänderten Achaten in Raum und Zeit und in Abhängigkeit vom Klima. Der Aufschluss 2006, 57, 129–150. [Google Scholar]
- Petránek, J. Sedimentäre Achate. Der Aufschluss 2009, 60, 291–302. [Google Scholar]
- Colburn, R.P. The Formation of Thundereggs; Geode Kid Productions: Deming, NM, USA, 2008. [Google Scholar]
- Dumańska-Słowik, M.; Natkaniec-Nowak, L.; Kotarba, M.J.; Sikorska, M.; Rzymełka, J.A.; Łoboda, A.; Gaweł, A. Mineralogical and geochemical characterization of the “bituminous” agates from Nowy Kościół (Lower Silesia, Poland). Neues Jahrbuch für Mineralogie Abhandlungen 2008, 184, 255–268. [Google Scholar]
- Dumańska-Słowik, M.; Natkaniec-Nowak, L.; Weselucha-Birczyńska, A.; Gaweł, A.; Lankosz, M.; Wróbel, P. Agates from Sidi Rahal, in the Atlas Mountains of Morocco: Gemmological characteristics and proposed origin. Gems Gemol. 2013, 49, 148–159. [Google Scholar]
- Dumańska-Słowik, M.; Powolny, T.; Sikorska-Jaworowska, M.; Gaweł, A.; Kogut, L.; Poloński, K. Characteristics and origin of agates from Płóczki Górne (Lower Silesia, Poland): A combined microscopic, micro-Raman, and cathodoluminescence study. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2018, 192, 6–15. [Google Scholar]
- Clark, R. South. Dakota’s State Gemstone—Fairburn Agate; Silverwind Agates: Appleton, CT, USA, 2009; 130p. [Google Scholar]
- Lyashenko, E.A. Agates of Russia. Mineral. Alm. 2010, 15, 6–27. [Google Scholar]
- Götze, J. Agate–Fascination between Legend and Science. In Agates III; Zenz, J., Ed.; Bode-Verlag: Haltern, Germany, 2011; pp. 19–133. [Google Scholar]
- Hurst, J.T. Dryhead Agate; Agate Treasures-Schatzkammerachate Publishing: Boulder, CO, USA, 2012; 90p. [Google Scholar]
- French, M.W.; Worden, R.H.; Lee, D.R. Electron backscatter diffraction investigation of length-fast chalcedony in agate: Implications for agate genesis and growth mechanisms. Geofluids 2013, 13, 32–44. [Google Scholar] [CrossRef]
- Richter, S.; Götze, J.; Niemeyer, H.; Möckel, R. Mineralogical investigation of agates from Cordón de Lila, Chile. J. Andean Geol. 2015, 42, 386–396. [Google Scholar]
- Ottens, B.; Götze, J. Achatwelt China; extralapis 51; Christian Weise Verlag: München, Germany, 2016; 110p. [Google Scholar]
- Natkaniec-Nowak, L.; Dumańska-Słowik, M.; Pršek, J.; Lankosz, M.; Wróbel, P.; Gaweł, A.; Kowalczyk, J.; Kocemba, J. Agates from Kerrouchen (The Atlas Mountains, Morocco). Minerals 2016, 6, 77. [Google Scholar] [CrossRef]
- Powolny, T.; Dumańska-Słowik, M.; Sikorska-Jaworowska, M.; Wójcik-Bania, M. Agate mineralization in spilitized Permian volcanics from “Borówno” quarry (Lower Silesia, Poland)—Microtextural, mineralogical, and geochemical constraints. Ore Geol. Rev. 2019, 114, 103–130. [Google Scholar] [CrossRef]
- Kigai, I.N. The genesis of agates and amethyst geodes. Can. Mineral. 2019, 57, 867–883. [Google Scholar] [CrossRef]
- Howard, C.B.; Rabinovitch, A. A new model of agate geode formation based on a combination of morphological features and silica sol-gel experiments. Eur. J. Mineral. 2017, 30, 97–106. [Google Scholar] [CrossRef]
- Gliozzo, E.; Cairncross, B.; Vennemann, T. A geochemical and micro-textural comparison of basalt-hosted chalcedony from the Jurassic Drakensberg and Neoarchean Ventersdorp Supergroup (Vaal River alluvial gravels), South Africa. Int. J. Earth Sci. 2019, 108, 1857–1877. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, L.; He, X. Gemological characteristics and origin of the Zhanguohong agate from Beipiao, Liaoning province, China: A combined microscopic, X-ray diffraction, and Raman spectroscopic study. Minerals 2020, 10, 401. [Google Scholar] [CrossRef]
- Pršek, J.; Dumańska-Słowik, M.; Powolny, T.; Natkaniec-Nowak, L.; Toboła, T.; Zych, D.; Skrepnicka, D. Agates from Western Atlas (Morocco)—Constraints from mineralogical and microtextural characteristics. Minerals 2020, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Moxon, T.; Carpenter, M.A. Crystallite growth kinetics in nanocrystalline quartz (agate and chalcedony). Mineral. Mag. 2009, 73, 551–568. [Google Scholar] [CrossRef]
- Walger, E. Das Vorkommen von Uruguay-Achaten bei Flonheim in Rheinhessen, seine tektonische Auswertung und seine Bedeutung für die Frage nach der Achatbildung. Jahresber. Mitt. Oberrh. Geol. Ver. 1954, 36, 20–31. [Google Scholar] [CrossRef]
- Holzhey, G. Herkunft und Akkumulation des SiO2 in Rhyolithkugeln aus Rotliegendvulkaniten des Thüringer Waldes. Geowiss. Mitt. Thüringen 1995, 3, 31–59. [Google Scholar]
- Hopkinson, L.; Roberts, S.; Herrington, R.; Wilkinson, J. Self-organization of submarine hydrothermal siliceous deposits: Evidence from the TAG hydrothermal mound, 26°N Mid-Atlantic Ridge. Geology 1998, 26, 347–350. [Google Scholar] [CrossRef]
- Taut, T.; Kleeberg, R.; Bergmann, J. Seifert software: The new Seifert Rietveld program BGMN and its application to quantitative phase analysis. Mater. Struct. 1998, 5, 57–66. [Google Scholar]
- Neuser, R.D.; Bruhn, F.; Götze, J.; Habermann, D.; Richter, D.K. Kathodolumineszenz: Methodik und Anwendung. Zentralblatt für Geologie und Paläontologie Teil I 1995, 1, 287–306. [Google Scholar]
- Götze, J.; Kempe, U. A comparison of optical microscope (OM) and scanning electron microscope (SEM) based cathodoluminescence (CL) imaging and spectroscopy applied to geosciences. Mineral. Mag. 2008, 72, 909–924. [Google Scholar] [CrossRef]
- Götze, J.; Pan, Y.; Stevens-Kalceff, M.; Kempe, U.; Müller, A. Origin and significance of the yellow cathodoluminescence (CL) of quartz. Am. Mineral. 2015, 100, 1469–1482. [Google Scholar] [CrossRef]
- Möckel, R.; Götze, J.; Sergeev, S.A.; Kapitonov, I.N.; Adamskaya, E.V.; Goltsin, N.A.; Vennemann, T. Trace-element analysis by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS): A case study for agates from Nowy Kościoł, Poland. J. Sib. Federal Univ. Eng. Technol. 2009, 2, 123–138. [Google Scholar]
- Flem, B.; Müller, A. In situ analysis of trace elements in quartz using Laser ablation inductively coupled plasma mass spectrometry. In Quartz: Deposits, Mineralogy and Analytics; Götze, J., Möckel, R., Eds.; Springer Geology: Heidelberg, Germany, 2012; pp. 219–236. [Google Scholar]
- Monecke, T.; Bombach, G.; Klemm, W.; Kempe, U.; Götze, J.; Wolf, D. Determination of trace elements in quartz standard UNS-SpS and in natural quartz by ICP-MS. Geostand. Newsl. 2000, 24, 73–81. [Google Scholar] [CrossRef]
- Venneman, T.W.; Morlok, A.; von Engelhardt, W.; Kyser, K. Stable isotope composition of impact glasses from the Nördlinger Ries impact crater, Germany. Geochim. Cosmochim. Acta 2001, 65, 1325–1336. [Google Scholar] [CrossRef]
- Richter-Feig, J.; Möckel, R.; Götze, J.; Heide, G. Investigation of fluids in chalcedony/quartz of agates using Thermogravimetry-Mass-Spectrometry. Minerals 2018, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Spötl, C.; Vennemann, T.W. Continuous-flow isotope ratio mass spectrometric analysis of carbonate minerals. Rapid Commun. Mass Spectrom. 2003, 17, 1004–1006. [Google Scholar] [CrossRef]
- Moxon, T.; Nelson, D.R.; Zhang, M. Agate recrystallization: Evidence from samples found in Archaean and Proterozoic host rocks, Western Australia. Aust. J. Earth Sci. 2008, 53, 235–248. [Google Scholar] [CrossRef]
- Gilg, H.A.; Morteani, G.; Kostitsyn, Y.; Preinfalk, C.; Gatter, I.; Strieder, A.J. Genesis of amethyst geodes in basaltic rocks of the Serra Geral Formation (Ametista do Sul, Rio Grande do Sul, Brazil): A fluid inclusion, REE, oxygen, carbon, and Sr isotope study on basalt, quartz, and calcite. Miner. Depos. 2003, 38, 1009–1025. [Google Scholar] [CrossRef]
- Gilg, H.A.; Krüger, Y.; Taubald, H.; van den Kerkhof, A.M.; Frenz, M.; Morteani, G. Mineralisation of amethyst-bearing geodes in Ametista do Sul (Brazil) from low-temperature sedimentary brines: Evidence from monophase liquid inclusions and stable isotopes. Miner. Depos. 2014, 49, 861–877. [Google Scholar] [CrossRef]
- Ottens, B.; Götze, J.; Schuster, R.; Krenn, K.; Hauzenberger, C.; Zsolt, B.; Vennemann, T. Exceptional multi-stage mineralization of secondary minerals in cavities of flood basalts from the Deccan Volcanic Province, India. Minerals 2019, 1019, 351. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.H. Chalcedony and quartz crystals in silicified coral. Am. Mineral. 1960, 45, 1304–1307. [Google Scholar]
- Rössler, R. Der Versteinerte Wald Von Chemnitz; Museum für Naturkunde Chemnitz: Chemnitz, Germany, 2001; 252p. [Google Scholar]
- Rich, P.V.; Rich, T.H.; Fenton, M.A.; Fenton, C.L. The Fossil Book—A Record of Prehistoric Life; Dover Publications: Mineola, NY, USA, 2020; 740p. [Google Scholar]
- Graetsch, H. Structural characteristics of opaline and microcrystalline silica minerals. Silica Rev. Mineral. 1994, 29, 209–232. [Google Scholar]
- Brewster, D. Über die Ursachen der Farben des irisierenden Achats. Ann. Phys. und Chem. 1844, 137, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Braitsch, O. Über die natürlichen Faser- und Aggregationstypen beim SiO2, ihre Verwachsungsformen, Richtungsstatistik und Doppelbrechung. Heidelb. Beiträge Mineral. Petrogr. 1957, 5, 331–372. [Google Scholar]
- Lange, P.; Blankenburg, H.-J.; Schrön, W. Rasterelektronenmikroskopische Untersuchungen an Vulkanitachaten. Z. Geol. Wiss. 1984, 12, 667–681. [Google Scholar]
- Miehe, G.; Graetsch, H.; Flörke, O.W. Crystal structure and growth fabric of length-fast chalcedony. Phys. Chem. Miner. 1984, 10, 197–199. [Google Scholar] [CrossRef]
- Folk, R.L.; Pittman, J.S. Length-slow chalcedony; A new testament for vanished evaporates. J. Sediment. Petrol. 1971, 41, 1045–1058. [Google Scholar]
- Flörke, O.W.; Jones, J.B.; Schmincke, H.-U. A new microcrystalline silica from Gran Canaria. Z. Krist. 1976, 143, 156–165. [Google Scholar] [CrossRef]
- Heaney, P.J.; Post, J.E. The widespread distribution of a novel silica polymorph in microcrystalline quartz varieties. Science 1992, 255, 441–443. [Google Scholar] [CrossRef]
- Heaney, P.J.; Veblen, D.R.; Post, J.E. Structural disparities between chalcedony and macrocrystalline quartz. Am. Mineral. 1994, 79, 452–460. [Google Scholar]
- Kingma, K.J.; Hemley, R.J. Raman spectroscopic study of microcrystalline silica. Am. Mineral. 1994, 79, 269–273. [Google Scholar]
- Nasdala, L.; Götze, J.; Gaft, M.; Hanchar, J.; Krbetschek, M. Luminescence techniques in Earth Sciences. In EMU Notes in Mineralogy; Beran, A., Libowitzky, E., Eds.; Eötvös University Press: Budapest, Hungary, 2004; Volume 6, pp. 1–49. [Google Scholar]
- Correns, C.W.; Nagelschmidt, G. Über Faserbau und optische Eigenschaften von Chalcedon. Z. für Krist. A 1933, 85, 199–213. [Google Scholar]
- Hoffmann, S. Untersuchungen über den Opalgehalt der Achate; Schweizerbart: Stuttgart, Germany, 1942; pp. 238–276. [Google Scholar]
- Flörke, O.W. Untersuchungen an amorphem und mikrokristallinem SiO2. Chem. der Erde 1962, 22, 91–110. [Google Scholar]
- Jones, B.; Renaut, R.W. Microstructural changes accompanying the opal-A to opal-CT transition: New evidence from the siliceous sinters of Geysir, Haukadalur, Iceland. Sedimentology 2007, 54, 921–948. [Google Scholar] [CrossRef]
- Moxon, T.; Reed, S.J.B. Agate and chalcedony from igneous and sedimentary hosts aged from 13 to 3480 Ma: A cathodoluminescence study. Mineral. Mag. 2006, 70, 485–498. [Google Scholar] [CrossRef]
- Moxon, T.; Reed, S.J.B.; Zhang, M. Metamorphic effects on agate found near the Shap granite, Cumbria, England: As demonstrated by petrography, X-ray diffraction and spectroscopic methods. Mineral. Mag. 2007, 71, 461–476. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry; John Wiley & Sons: New York, NY, USA, 1979; p. 866. [Google Scholar]
- Holzhey, G. Mikrokristalline SiO2-Mineralisationen in rhyolithischen Rotliegendvulkaniten des Thüringer Waldes (Deutschland) und ihre Genese. Chem. der Erde 1999, 59, 183–205. [Google Scholar]
- Tennyson, C. “Struktur” und Farbenspiel des Edelopals. Lapis 1979, 4, 13–15. [Google Scholar]
- Dong, G.; Morrison, G.; Jaireth, S. Quartz textures in epithermal veins, Queensland—Classification, origin, and implication. Econ. Geol. 1995, 90, 1841–1856. [Google Scholar] [CrossRef]
- Weil, J.A. A review of electron spin spectroscopy and its application to the study of paramagnetic defects in crystalline quartz. Phys. Chem. Miner. 1984, 10, 149–165. [Google Scholar] [CrossRef]
- Weil, J.A. A review of the EPR spectroscopy of the point defects in α-quartz: The decade 1982–1992. In Physics and Chemistry of SiO2 and the Si-SiO Interface 2; Helms, C.R., Deal, B.E., Eds.; Plenum Press: New York, NY, USA, 1993; pp. 131–144. [Google Scholar]
- Stevens-Kalceff, M.A. Cathodoluminescence microcharacterization of point defects in a-quartz. Mineral. Mag. 2009, 73, 585–606. [Google Scholar] [CrossRef]
- Götze, J. Chemistry, textures and physical properties of quartz—Geological interpretation and technical application. Mineral. Mag. 2009, 73, 645–671. [Google Scholar] [CrossRef]
- Götze, J.; Plötze, M.; Fuchs, H.; Habermann, D. Defect structure and luminescence behaviour of agate—Results of electron paramagnetic resonance (EPR) and cathodoluminescence (CL) studies. Mineral. Mag. 1999, 63, 149–163. [Google Scholar] [CrossRef]
- Mashkovtsev, R.I.; Li, Z.; Mao, M.; Pan, Y. 73Ge, 17O and 29Si hyperfine interactions of the Ge E′1 center in crystalline SiO2. J. Magn. Reson. 2013, 233, 7–16. [Google Scholar] [CrossRef]
- SivaRamaiah, G.; Lin, J.R.; Pan, Y. Electron paramagnetic resonance spectroscopy of Fe3+ ions in amethyst: Thermodynamic potentials and magnetic susceptibility. Phys. Chem. Miner. 2011, 38, 159–167. [Google Scholar] [CrossRef]
- Pan, Y.; Hu, B. Radiation-induced defects in quartz. IV. Thermal properties and implications. Phys. Chem. Miner. 2009, 36, 421–430. [Google Scholar] [CrossRef]
- Botis, S.; Nokhrin, S.M.; Pan, Y.; Xu, Y.; Bonli, T. Natural radiation-induced damage in quartz. I. Correlations between cathodoluminescence colors and paramagnetic defects. Can. Mineral. 2005, 43, 1565–1580. [Google Scholar] [CrossRef] [Green Version]
- Nilges, M.J.; Pan, Y.; Mashkovtsev, R.I. Radiation-induced defects in quartz. I. Single-crystal W-band EPR study of an electron irradiated quartz. Phys. Chem. Miner. 2008, 35, 103–115. [Google Scholar] [CrossRef]
- Nilges, M.J.; Pan, Y.; Mashkovtsev, R.I. Radiation-induced defects in quartz. III. EPR, ENDOR and ESEEM characterization of a peroxy radical. Phys. Chem. Miner. 2009, 36, 63–71. [Google Scholar] [CrossRef]
- Pan, Y.; Nilges, M.J.; Mashkovtsev, R.I. Radiation-induced defects in quartz. II. W-band single-crystal EPR study of natural citrine. Phys. Chem. Miner. 2008, 35, 387–397. [Google Scholar] [CrossRef]
- Pan, Y.; Nilges, M.J.; Mashkovtsev, R.I. Multifrequency single-crystal EPR characterization and DFT modeling of new peroxy radicals in quartz. Mineral. Mag. 2009, 73, 517–535. [Google Scholar] [CrossRef]
- Walsby, C.J.; Lees, N.S.; Claridge, R.F.C.; Weil, J.A. The magnetic properties of oxygen-hole aluminum centres in crystalline SiO2. VI: A stable AlO4/Li centre. Can. J. Phys. 2003, 81, 583–598. [Google Scholar] [CrossRef]
- Götze, J. Application of cathodoluminescence (CL) microscopy and spectroscopy in geosciences. Microsc. Microanal. 2012, 18, 1270–1284. [Google Scholar] [CrossRef] [Green Version]
- Götze, J.; Plötze, M.; Habermann, D. Cathodoluminescence (CL) of quartz: Origin, spectral characteristics and practical applications. Mineral. Petrol. 2001, 71, 225–250. [Google Scholar] [CrossRef]
- Götze, J.; Hanchar, J. Atlas of Cathodoluminescence (CL) Microtextures; GAC Miscellaneous Publication No. 10; Geological Association of Canada: St. John’s, NL, Canada, 2018; 248p.
- Ramseyer, K.; Baumann, J.; Matter, A.; Mullis, J. Cathodoluminescence colours of alpha-quartz. Mineral. Mag. 1988, 52, 669–677. [Google Scholar] [CrossRef]
- Siegel, G.H.; Marrone, M.J. Photoluminescence in as-drawn and irradiated silica optical fibers: An assessment of the role of non-bridging oxygen defect centres. J. Non-Cryst. Solids 1981, 45, 235–247. [Google Scholar] [CrossRef]
- Alonso, P.J.; Halliburton, L.E.; Kohnke, E.E.; Bossoli, R.B. X-ray induced luminescence in crystalline SiO2. J. Appl. Phys. 1983, 54, 5369–5375. [Google Scholar] [CrossRef]
- Luff, B.J.; Townsend, P.D. Cathodoluminescence of synthetic quartz. J. Phys. Condens. Matter 1990, 2, 8089–8097. [Google Scholar] [CrossRef]
- Perny, B.; Eberhardt, P.; Ramseyer, K.; Mullis, J. Microdistribution of aluminium, lithium and sodium in quartz: Possible causes and correlation with short-lived cathodoluminescence. Am. Mineral. 1992, 77, 534–544. [Google Scholar]
- Götze, J.; Plötze, M.; Graupner, T.; Hallbauer, D.K.; Bray, C. Trace element incorporation into quartz: A combined study by ICP-MS, electron spin resonance, cathodoluminescence, capillary ion analysis and gas chromatography. Geochim. Cosmochim. Acta 2004, 68, 3741–3759. [Google Scholar] [CrossRef]
- Blankenburg, H.-J.; Schrön, W. Zum Spurenelementchemismus der Vulkanitachate. Chem. Erde 1982, 41, 121–135. [Google Scholar]
- Mason, B. Cosmochemistry, Part. I. Meteorites. In Data of Geochemistry; Fleischer, M., Ed.; U.S. Geological Survey Professional Paper: Reston, VA, USA, 1979. [Google Scholar]
- Walenzak, Z. Geochemistry of minor elements dispersed in quartz (Ge, Al, Ga, Fe, Ti, Li and Be). Arch. Mineral. 1969, 28, 189–335. [Google Scholar]
- Konstatinov, W.M. Uranium bearing lithophysae in acidic extrusive rocks. Izvestiya Akademii Nauk SSSR Seria Geologika 1968, 7, 43–49. (In Russian) [Google Scholar]
- Zielinski, R.A. Uranium mobility during interaction of rhyolitic obsidian, perlite and felsite with alkaline carbonate solution: T = 120 °C, P = 210 kg/cm2. Chem. Geol. 1979, 27, 47–63. [Google Scholar] [CrossRef]
- Pan, Y.; Li, D.; Feng, R.; Wiens, E.; Chen, N.; Götze, J.; Lin, J. Uranyl binding mechanism in microcrystalline silicas: A potential missing link for uranium mineralization by direct uranyl co-precipitation and environmental implications. Geochim. Cosmochim. Acta 2021, 292, 518–531. [Google Scholar] [CrossRef]
- Peppard, D.F.; Mason, G.W.; Lewey, S. A tetrad effect in the liquid–liquid extraction ordering of lanthanide(III). J. Inorg. Nucl. Chem. 1969, 31, 2271–2272. [Google Scholar] [CrossRef]
- Monecke, T.; Dulski, P.; Kempe, U. Origin of convex tetrads in rare earth element patterns of hydrothermally altered siliceous igneous rocks from the Zinnwald Sn–W deposit, Germany. Geochem. Cosmochim. Acta 2007, 71, 335–353. [Google Scholar] [CrossRef]
- Wood, S.A. The aqueous geochemistry of the rare-earth elements and yttrium. 2. Theoretical predictions of speciation in hydrothermal solutions to 350 °C at saturation water vapor pressure. Chem. Geol. 1990, 88, 99–125. [Google Scholar] [CrossRef]
- Kempe, U.; Götze, J.; Belyatsky, B.V.; Plötze, M. Ce anomalies in monazite, fluorite and agate from Permian volcanics of the Saxothuringian (Germany). J. Czech. Geol. Soc. 1998, 42, 38. [Google Scholar]
- Barsanov, G.P.; Plyusnina, I.I.; Jakovleva, M.E. Specific features of the chemical composition, physical properties and the structure of chalcedony. In New Data of Minerals; Nauka: Moscow, Russia, 1981; Volume 28, pp. 3–33. (In Russian) [Google Scholar]
- Hoefs, J. Stable Isotope Geochemistry, 4th ed.; Springer: Heidelberg, Germany, 1997; 201p. [Google Scholar]
- Ingerson, E.; Weshow, R.L. Oxygen isotope fractionation in the system quartz–water. Geochem. Int. 1965, 2, 691–707. [Google Scholar]
- Matsuhisa, Y.; Goldsmith, J.R.; Clayton, R.N. Oxygen isotopic fractionation in the system quarz-albite–anorthite-water. Geochim. Cosmochim. Acta 1979, 43, 1131–1140. [Google Scholar] [CrossRef]
- Harris, C. Oxygen-isotope zonation of agates from Karoo volcanics of the Skeleton Coast, Namibia. Am. Mineral. 1989, 74, 476–481. [Google Scholar]
- Strauch, G.; Nitzsche, H.-M.; Holzhey, G. Isotopenuntersuchungen an Rhyolithen und Achatbildungen. Neues Jahrb. für Mineral. Abh. 1994, 165, 103–104. [Google Scholar]
- Blankenburg, H.J.; Pilot, J.; Werner, C.D. Erste Ergebnisse der Sauerstoffisotopenuntersuchungen an Vulkanitachaten und ihre genetische Interpretation. Chem. der Erde 1982, 41, 213–217. [Google Scholar]
- Harris, C. Oxygen isotope geochemistry of a quartz-agate geode from north-western Namibia. Commun. Geol. Surv. S.W. Afr. Namib. 1988, 4, 43–44. [Google Scholar]
- Holzhey, G. Contribution to petrochemical-mineralogical characterization of alteration processes within the marginal facies of rhyolitic volcanics of lower Permian Age, Thuringian Forest, Germany. Chem. Erde 2001, 61, 149–186. [Google Scholar]
- Fallick, A.E.; Jocely, J.; Hamilton, P.J. Oxygen and hydrogen stable isotope systematics in Brazilian agates. In Geochemistry and Mineral Formation in the Earth Surface; Rodriguez-Clemente, R., Tardy, Y., Eds.; Editorial CSIC: Madrid, Spain, 1987; pp. 99–117. [Google Scholar]
- Duarte, L.C.; Hartmann, L.A.; Ronchi, L.H.; Berner, Z.; Theye, T.; Massonne, H.J. Stable isotope and mineralogical investigation of the genesis of amethyst geodes in the Los Catalanes gemological district, Uruguay, southernmost Paraná volcanic province. Miner. Depos. 2011, 46, 239–255. [Google Scholar] [CrossRef]
- Rezaei-Kahkhaei, M.; Ansarifar, O.; Ghasemi, H. Geochemistry and oxygen stable isotopes of Reza Abad agates, SE Shahrood, Central Iran: An approach to temperature and formation process. J. Econ. Geol. 2019, 11, 3. [Google Scholar]
- Kita, I.; Taguchi, S. Oxygen isotopic behaviour of precipitating silica from geothermal water. Geochem. J. 1986, 20, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Blankenburg, H.-J.; Thomas, R.; Klemm, W.; Leeder, O. Interpretation der Ergebnisse von Einschlußunersuchungen an den Quarzinkrustaten aus Vulkanitachaten. Z. Geol. Wiss. 1990, 18, 81–85. [Google Scholar]
- Frondel, C. Structural hydroxyl in chalcedony (Type B quartz). Am. Mineral. 1982, 67, 1248–1257. [Google Scholar]
- Graetsch, H.; Flörke, O.W.; Miehe, G. The nature of water in chalcedony and opal-C from Brazilian agate geodes. Phys. Chem. Miner. 1985, 12, 300–306. [Google Scholar] [CrossRef]
- Thomas, R.; Blankenburg, H.-J. Erste Ergebnisse über Einschlussuntersuchungen an Quarzen aus Achatmandeln und –kugeln basischer und saurer Vulkanite. Z. Geol. Wiss. 1981, 9, 625–633. [Google Scholar]
- Commin-Fischer, A.; Berger, G.; Polvė, M.; Dubois, M.; Sardini, P.; Beaufort, D.; Formoso, M. Petrography and chemistry of SiO2 filling phases in the amethyst geodes from the Serra Geral Formation deposit, Rio Grande do Sul, Brazil. J. South. Am. Earth Sci. 2010, 29, 751–760. [Google Scholar] [CrossRef]
- Ramboz, C.; Pichavant, M.; Weisbrod, A. Fluid immiscibility in natural processes: Use and misuse of fluid inclusion data. Chem. Geol. 1982, 37, 29–48. [Google Scholar] [CrossRef]
- Roedder, E. Fluid Inclusions; Reviews in Mineralogy 12; Mineralogical Society of America: Reston, VA, USA, 1984; 645p. [Google Scholar]
- Hall, D.L.; Sterner, S.M.; Bodnar, R.J. Freezing point depression of NaCl–KCl–H2O solutions. Econ. Geol. 1988, 83, 197–202. [Google Scholar] [CrossRef]
- Tripp, R.B. The mineralogy of Warsaw Formation geodes. Iowa Acad. Sci. Proc. 1959, 66, 350–356. [Google Scholar]
- Hayes, J.B. Geodes and concretions from the Mississippian Warsaw Formation. Keokuk region, Iowa, Illinois, Missouri. Sediment. Petrol. 1964, 34, 123–133. [Google Scholar]
- Holzhey, G. Die Paragenese von Mineralen in Rhyolithkugeln (Lithophysen) aus Rotliegendvulkaniten des Thüringer Waldes. Semana 2016, 31, 25–50. [Google Scholar]
- Schmitt-Riegraf, C. Magmenentwicklung und spät- bis post-magmatische Alterationsprozesse permischer Vulkanite im Nordwesten der Nahe-Mulde. Münstersche Forsch. zur Geol. und Paläontologie 1996, 80, 1–251. [Google Scholar]
- Walger, E. Zur Entstehung des Calcitachates. Fortschr. Mineral. 1961, 39, 360. [Google Scholar]
- Landmesser, M. Calcitachat: Zur Deutung eines verblüffenden mineralogischen Phänomens. Mainz. Nat. Arch. 1996, 34, 9–43. [Google Scholar]
- Blankenburg, H.-J.; Unterricker, S.; Eichler, B.; Starke, R.; Stolz, W.; Rösler, H.J. Natürliche Radioaktivität, chemische und Phasenzusammensetzung der Eisenoxide aus Vulkanitachaten. Chem. Erde 1986, 45, 159–166. [Google Scholar]
- Rosemeyer, T. Copper-banded agates from the Kearsarge copper-bearing amygdaloidal lode Houghton county, Michigan. Rocks Miner. 2012, 87, 352–365. [Google Scholar] [CrossRef]
- Moxon, T. Agate in thin section. Rocks Miner. 2014, 89, 328–339. [Google Scholar] [CrossRef]
- Breitkreuz, C. Spherulites and lithophysae—200 years of investigation on hightemperature crystallization domains in silica-rich volcanic rocks. Bull. Volcanol. 2013, 75, 705–720. [Google Scholar] [CrossRef]
- Sakka, S.; MacKenzie, J.D. Relationship between apparent glass transition temperature and liquidus temperature for inorganic glasses. J. Non-Cryst. Solids 1971, 6, 145–162. [Google Scholar] [CrossRef]
- Lofgren, G. Spherulitic textures in glassy and crystalline rocks. J. Geophys. Res. 1971, 76, 5635–5648. [Google Scholar] [CrossRef]
- Breitkreuz, C.; Götze, J.; Weißmantel, A. Mineralogical and geochemical investigation of megaspherulites from Argentina, Germany and USA. Bull. Volcanol. 2020. accepted for publication. [Google Scholar]
- Herrington, R.J.; Wilkinson, J.J. Colloidal gold and silica in mesothermal vein systems. Geology 1993, 21, 539–542. [Google Scholar] [CrossRef]
- Jebrag, M. Hydrothermal breccias in vein-type ore deposits: A review of mechanisms, morphology and size distribution. Ore Geol. Rev. 1997, 12, 111–134. [Google Scholar] [CrossRef]
- Haake, R.; Fischer, J.; Reißmann, R. Über das Achat-Amethyst-Vorkommen von Schlottwitz im Osterzgebirge. Mineralienwelt 1991, 2, 20–24. [Google Scholar]
- Chowns, T.M.; Elkins, J.E. The origin of quartz geodes and cauliflower cherts through the silification of anhydrite nodules. J. Sediment. Petrol. 1974, 44, 885–903. [Google Scholar]
- Tucker, M.E. Quartz replaced anhydrite nodules (“Bristol diamonds”) from the Triassic of the Bristol District. Geol. Mag. 1976, 113, 569–574. [Google Scholar] [CrossRef]
- Jacka, A.D. Replacement of fossils by length-slow chalcedony and associated dolomitization. J. Sediment. Petrol. 1974, 44, 421–427. [Google Scholar]
- Landmesser, M. Zur Entstehung von Kieselhölzern. ExtraLapis 1994, 7, 49–80. [Google Scholar]
- Milliken, K.L. The silicified evaporate syndrome: Two aspects of silicification history of former evaporate nodules from Southern Kentucky and Northern Tennessee. J. Sediment. Petrol. 1979, 49, 245–256. [Google Scholar]
- Shaub, B.M. The Origin of Agates, Thundereggs, and Other Nodular Structures; Agate Publishing Company: Northampton, MA, USA, 1989; 105p. [Google Scholar]
- Landmesser, M. Zur Geothermometrie und Theorie der Achate. Mitt. Pollichia 1992, 79, 159–201. [Google Scholar]
- Harder, H. Agates-formation as a multi component colloid chemical precipitation at low temperatures. Neues Jahrb. Mineral. Mon. 1993, H.1, 31–48. [Google Scholar]
- Williamson, B.J.; Wilkinson, J.J.; Luckham, P.F.; Stanley, C.J. Formation of coagulated colloidal silica in high-temperature mineralizing fluids. Mineral. Mag. 2002, 66, 547–553. [Google Scholar] [CrossRef]
- White, J.F.; Corwin, J.F. Synthesis and origin of chalcedony. Am. Mineral. 1961, 46, 112–119. [Google Scholar]
- Flörke, O.W. Transport and deposition of SiO2 with H2O under supercritical conditions. Krist. Tech. 1972, 7, 159–166. [Google Scholar] [CrossRef]
- Oehler, J.H. Hydrothermal crystallization of silica gel. Geol. Soc. America Bull. 1976, 87, 1143–1152. [Google Scholar] [CrossRef]
- Götze, J.; Plötze, M.; Tichomirowa, M.; Fuchs, H.; Pilot, J. Aluminium in quartz as an indicator of the temperature of formation of agate. Mineral. Mag. 2001, 65, 407–413. [Google Scholar] [CrossRef]
- Dennen, W.H.; Blackburn, W.H.; Quesada, A. Aluminum in quartz as a geothermometer. Contrib. Mineral. Petrol. 1970, 27, 332–342. [Google Scholar] [CrossRef]
- Agel, A.; Petrov, I. Im Quarzgitter substituiertes Aluminium als Indikator für dessen Bildungstemperatur. Eur. J. Miner. 1990, 2, 144. [Google Scholar]
- Correns, C.W. The experimental weathering of silicates. Clay Miner. Bull. 1961, 4, 249–265. [Google Scholar] [CrossRef]
- Wirsching, U. Experimente zum Einfluß des Gesteinsglas-Chemismus auf die Zeolithbildung durch hydrothermale Umwandlung. Contrib. Mineral. Petrol. 1975, 49, 117–124. [Google Scholar] [CrossRef]
- Seyfried, W.E.; Bischo, J.L. Low temperature basalt alteration by seawater: An experimental study at 70 °C and 150 °C. Geochim. Cosmochim. Acta 1979, 43, 1937–1947. [Google Scholar] [CrossRef]
- Giggenbach, W.F. Mass transfer in hydrothermal alteration systems—A conceptual approach. Geochim. Cosmochim. Acta 1984, 48, 2693–2711. [Google Scholar] [CrossRef]
- Duplay, J.; Paquet, H.; Kossovskaya, A.; Tard, Y. Estimation de la température de formation des paragenèses saponite-céladonite et glauconite-nontronite dans les altérations sous-marines de basalte, par la méthode des corrélations entre éléments au sein de populations monominérales. CR Acad. Sci. Paris 1989, 309, 53–58. [Google Scholar]
- Klammer, D. Mass change during extreme acid-sulphate hydrothermal alteration of a Tertiary latite, Styria, Austria. Chem. Geol. 1997, 141, 33–48. [Google Scholar] [CrossRef]
- Polgári, M.; Hein, J.R.; Németh, T.; Pál-Molnár, E.; Vigh, T. Celadonite and smectite formation in the Úrkút Mn-carbonate ore deposit (Hungary). Sediment. Geol. 2013, 294, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Götze, J.; Hofmann, B.; Machałowski, T.; Tsurkan, M.V.; Jesionowski, T.; Ehrlich, H.; Kleeberg, R.; Ottens, B. Biosignatures in subsurface filamentous fabrics (SFF) from the Deccan Volcanic Province, India. Minerals 2020, 10, 540. [Google Scholar] [CrossRef]
- Sukheswala, R.N.; Avasia, R.K.; Gangopadhyay, M. Zeolites and associated secondary minerals in the Deccan Traps of western India. Mineral. Mag. 1974, 39, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Mattioli, M.; Cenni, M.; Passaglia, E. Secondary mineral assemblages as indicators of multistage alteration processes in basaltic lava flows: Evidence from the Lessini Mountains, Veneto Volcanic Province, Northern Italy. Period. Di Mineral. 2016, 85, 1–24. [Google Scholar]
- Kryza, R. Bilans chemiczny dla stref mineralizacji agatowej w wulkanitach rejonu Nowego Kośioła (Góry Kaczawskie). Arch. Mineral. 1983, 39, 125–132. [Google Scholar]
- Krauskopf, K.B. Dissolution and precipitation of silica at low temperatures. Geochim. Cosmochim. Acta 1956, 10, 1–26. [Google Scholar] [CrossRef]
- Landmesser, M. Mobility by metastability: Silica transport and accumulation at low temperatures. Chem. Erde 1995, 55, 149–176. [Google Scholar]
- Dietzel, M. Dissolution of silicates and the stability of polysilicic acid. Geochim. Et Cosmochim. Acta 2000, 64, 3275–3281. [Google Scholar] [CrossRef]
- Gránásy, L.; Pusztai, T.; Tegze, G.; Warren, J.A.; Douglas, J.F. Growth and form of spherulites. Phys. Rev. 2005, 72, 11605–11619. [Google Scholar] [CrossRef] [Green Version]
- Krug, H.-J.; Jacob, K.-H. Genese und Fragmentierung rhytmischer Bänderungen durch Selbstorganisation. Z. Dtsch. Geol. Ges. 1994, 144, 452–460. [Google Scholar]
- Bryxina, N.A.; Sheplev, V.S. Auto-oscillation in agate crystallization. Math. Geol. 1999, 31, 297–309. [Google Scholar] [CrossRef]
- Wang, Y.; Merino, E. Self-organizational origin of agates: Banding, fiber twisting, composition, and dynamic crystallizationmodel. Geochim. Cosmochim. Acta 1990, 54, 1627–1638. [Google Scholar] [CrossRef]
- Götze, J.; Stanek, K.; Orozco, G. Auf Achatsuche in Kuba. Mineralienwelt 2020, 3, 56–63. [Google Scholar]
- Goldbaum, J.; Howard, C.; Rabinovitch, A. Spatial Chirp of agate bands. Minerals 2019, 9, 634. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, A.S.; Showalter, K. Control of waves, patterns and turbulence in chemical systems. Phys. Rep. 2006, 425, 79–194. [Google Scholar] [CrossRef]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallisation: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Revs. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [Green Version]
- Papineau, D. Chemically oscillation reactions in the formation of botryoidal malachite. Am. Mineral. 2020, 105, 447–454. [Google Scholar] [CrossRef]
- Walger, E.; Mattheß, G.; Von Seckendorff, V.; Liebau, F. The formation of agate structures: Models for silica transport, agate layer accretion, and for flow patterns and flow regimes in infiltration channels. Neues Jahrb. Mineral. Abh. 2009, 186, 113–152. [Google Scholar] [CrossRef]
- Laurs, B.M.; Rossman, G.R. Grape-like “Manakarra” quartz from Sulawesi, Indonesia. J. Gemmol. 2018, 36, 101–102. [Google Scholar]
- Hofmann, B.A. Subsurface filamentous fabrics. In Encyclopedia of Geobiology; Reitner, J., Thiel, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 851–853. [Google Scholar]
- Thewalt, U.; Dörfner, G. Wie kommt das Moos in den Achat und wie nicht? Der Aufschluss 2012, 63, 1–16. [Google Scholar]
- Reysenbach, A.-L.; Cady, S.L. Microbiology of ancient and modern hydrothermal systems. Trends Microbiol. 2001, 9, 79–86. [Google Scholar] [CrossRef]
- Jones, B.; Renault, R.W. Influence of thermophilic bacteria on calcite and silica precipitation in hot springs with water temperatures above 90 °C: Evidence from Kenya and New Zealand. Can. J. Earth Sci. 1996, 33, 72–83. [Google Scholar] [CrossRef]
- Konhauser, K.O.; Phoenix, V.R.; Bottrell, S.H.; Adams, D.G.; Head, I.M. Microbial-silica interaction in Icelandic hot spring sinter: Possible analogues for some Precambrian siliceous stromatolites. Sedimentology 2001, 48, 415–433. [Google Scholar] [CrossRef]
- Ferris, F.G.; Beveridge, T.J.; Fyfe, W.S. Iron-silica crystallite nucleation by bacteria in a geothermal sediment. Nature 1986, 320, 609–611. [Google Scholar] [CrossRef]
- Parenteau, M.N.; Cady, S.L. Microbial biosignatures in iron-mineralized phototrophic mats at Chocolate Pots Hot Springs, Yellowstone National Park, United States. PALAIOS 2010, 25, 97–111. [Google Scholar] [CrossRef]
- Müller, A.; Polgári, M.; Gucsik, A.; Pál-Molnár, E.; Koós, M.; Veres, M.; Götze, J.; Nagy, S.; Cserháti, C.; Németh, T.; et al. Cathodoluminescent features and Raman spectroscopy of Miocene hydrothermal biomineralization embedded in cryptocrystalline silica varieties, Central Europe, Hungary. In Micro-Raman Spectroscopy and Luminescence Studies in the Earth and Planetary Sciences; Gucsik, A., Ed.; American Institute of Physics: Houston, TX, USA, 2009; pp. 207–218. [Google Scholar]
- Yang, H.; Coombs, N.; Ozin, G.A. Morphogenesis of shapes and surface patterns in mesoporous silica. Nature 1997, 386, 692–695. [Google Scholar] [CrossRef]
- Glaab, F.; Kellermeier, M.; Kunz, W.; Morallon, E.; Garcia-Ruiz, J.M. Formation and evolution of chemical gradients and potential differences across self-assembling inorganic membranes. Angew. Chem. 2012, 124, 4393–4397. [Google Scholar] [CrossRef]
- Moxon, T.; Petrone, C.M.; Reed, S.J.B. Characterization and genesis of horizontal banding in Brazilian agate: An X-ray diffraction, thermogravimetric and electron microprobe study. Mineral. Mag. 2013, 77, 227–248. [Google Scholar] [CrossRef] [Green Version]
- Haudin, F.; Cartwright, J.H.E.; Braua, F.; De Wit, A. Spiral precipitation patterns in confined chemical gardens. Proc. Natl. Acad. Sci. USA 2014, 111, 17363–17367. [Google Scholar] [CrossRef] [Green Version]
- Noorduin, W.L.; Grinthal, A.; Mahadevan, L.; Aizenberg, J. Rationally designed complex, hierarchical microarchitectures. Science 2013, 340, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Kellermeier, M.; Cölfen, H.; García-Ruiz, J.M. Silica Biomorphs: Complex biomimetic hybrid materials from “sand and chalk”. Eur. J. Inorg. Chem. 2012, 32, 5123–5144. [Google Scholar] [CrossRef]
- Dominguez-Bella, S.; Garcia-Ruiz, J.M. Textures in induced morphology crystal aggregates of CaCO3: Sheaf of wheat morphologies. J. Cryst. Growth 1986, 79, 236–240. [Google Scholar] [CrossRef]
- Lenz, G.; Schäfer, K. Spiralen im Achat—Ein biologisches oder ein mineralogisches Phänomen? Lapis 2008, 1, 21–24. [Google Scholar]



































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Agates in Acidic Volcanic Rocks | Agates in Basic Volcanic Rocks | Vein Agates | Agates in Sedimentary Rock | ||||
|---|---|---|---|---|---|---|---|---|
| Chalcedony | Quartz | Chalcedony | Quartz | Chalcedony | Quartz | Chalcedony | Quartz | |
| Al | 109…7300 | 34…1380 | 15…1170 | 30…700 | 640…7000 | 75…546 | 103…809 | 1…13 |
| Ca | <10…540 | <10…27 | <10…710 | <10…120 | 20…400 | <16…45 | 350…1400 | 630…1200 |
| Fe | 30…4500 | <30…978 | 7…3900 | <30…670 | 150…3000 | 150…1700 | 26…1600 | <1…30 |
| Mn | <1…120 | <1…10 | <1…90 | <1…46 | 1…27 | 3…6 | <1 | <1 |
| K | 40…3640 | 30…600 | 20…580 | 66…420 | 150…1360 | 25…480 | na | na |
| Na | 90…850 | 22…600 | 85…750 | 20…140 | 50…350 | 100…240 | 48…323 | 45…158 |
| Mg | 5…556 | 3…70 | 1…364 | 4…287 | 16…220 | 46…105 | na | na |
| F | <25…380 | <25…52 | <25…228 | <25…270 | 79…156 | 31…200 | na | na |
| Ge | 3.2…22 | 0.9…30 | 0.5…8.5 | 0.5…15 | 1.3…4.8 | 0.6…4.5 | <1…2 | <1 |
| B | <10…135 | <2…155 | 3…146 | <2…120 | <2…18 | 6…46 | na | na |
| U | <1…21 | <1…14 | <1…21 | <1…4 | <1…15 | <1…4 | <1…72 | 5…18 |
| Spot | Ge | Al | Fe | B | Ga | Ca | Li | Na | K | Rb |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 77.4 | - | 11.5 | 36 | 0.07 | - | 0.98 | 77.4 | 41.1 | 0.45 |
| 2 | 16.4 | 366 | 46.9 | 34.2 | 0.71 | 62.4 | 1.66 | 101 | 162 | 1.86 |
| 3 | 22.2 | 329 | 48.2 | 33.3 | 0.81 | 58.3 | 1.89 | 67.4 | 168 | 1.5 |
| 4 | 94.9 | - | 11.3 | 33.4 | 0.16 | - | 0.89 | - | 61.8 | 0.5 |
| 5 | 53.5 | - | 12.4 | 23.5 | 0.04 | - | 0.86 | - | 49.2 | 0.32 |
| 6 | 93.6 | - | 14.5 | 31.3 | 0.12 | - | 0.51 | - | 37.4 | 0.44 |
| 7 | 26.8 | - | 18.7 | 8 | 0.14 | - | 0.59 | - | 32 | 0.15 |
| 8 | 68.9 | - | 12.8 | 19.2 | 0.12 | - | 0.47 | - | 33 | 0.24 |
| 9 | 31.6 | 103 | 154 | 28.7 | 0.38 | 9.7 | 0.49 | 37.9 | 89.2 | 0.58 |
| 10 | 36.3 | <6.0 | 47.7 | 29.2 | 0.17 | 9.4 | 0.39 | - | 51.9 | 0.33 |
| 11 | 28.9 | - | 47.8 | 41 | 0.11 | - | 0.49 | - | 55.1 | 0.43 |
| 12 | 20.4 | - | 61.9 | 35.7 | 0.12 | - | 0.59 | - | 60.7 | 0.33 |
| 13 | 13.2 | <6.0 | 55.4 | 45.6 | 0.22 | - | 0.35 | - | 60.2 | 0.31 |
| 14 | 21.2 | 162 | 66.9 | 25.8 | 0.98 | - | 0.30 | - | 149 | 0.97 |
| 15 | 0.5 | - | 1.9 | <1.0 | 0.18 | 53.3 | 0.30 | - | 1.2 | - |
| Agate Location | Host Rock/Agate Type | Thom |
|---|---|---|
| Gehlberg (Thuringia) | rhyolite/lithophysa | 95 °C |
| Chemnitz (Saxony) | ignimbrite/lithophysa | 176 °C |
| St. Egidien (Saxony) | ignimbrite/lithophysa | 186 °C |
| Mügeln (Saxony) | rhyolite/lithophysa | 177 °C |
| Burgstall (Saxony) | rhyolite/lithophysa | 172 °C |
| Gröppendorf (Saxony) | melaphyre/amygdale | 134 °C |
| Schlottwitz (Saxony) | hydrothermal vein agate | 80 °C |
| Mineral Group | Minerals |
|---|---|
| Elements | copper, lead, sulphur, graphite |
| Sulfides | pyrite, marcasite, sphalerite, galenite, chalkopyrite, covelline |
| Oxides/Hydroxides | hematite, goethite |
| todorocite, ramsdellite, birnessite, pyrolusite, rancieite, hollandite, | |
| cryptomelane/psilomelane, manganite | |
| cuprite, rutile, anatase, spinel (magnesio-chromite) | |
| Carbonates | calcite, aragonite, dolomite, siderite, ankerite, rodochrosite, strontianite, magnesite, smithsonite, malachite, azurite, bastnesite-(Ce) |
| Sulfates | barite, celestine, anhydrite, gypsum |
| Phosphates | apatite, monazite-(Ce), rabdophane-(Ce), xenotime-(Y) |
| Halides | fluorite |
| Silicates | clay minerals (kaolinite, illite, montmorillonite, beidellite, saponite, nontronite) |
| zeolites (mordenite, heulandite/clinoptilolite, harmotome, chabasite, natrolite, analcime, scolezite, mesolite, stilbite, thomsonite, laumontite, brewsterite, philipsite) | |
| glauconite–celadonite series, chlorite, serpentine, talc, prehnite, | |
| datolithe, epidote, apophyllite, chrysocolle, orthoclase, albite, plagioclase |
| Phase | Pitchstone | Altered Pitchstone |
|---|---|---|
| Amorphous (glass) | 63.7 ± 2.2 | - |
| Plagioclase | 16.1 ± 0.9 | 14.3 ± 1.0 |
| K-feldspar | 5.5 ± 0.5 | 8.7 ± 0.9 |
| Quartz | 1.7 ± 0.2 | 9.6 ± 0.7 |
| Biotite | 1.1 ± 0.9 | - |
| Magnetite | 0.8 ± 0.2 | - |
| Ilmenite | 0.5 ± 0.2 | - |
| Montmorillonite | 10.6 ± 1.9 | 62.0 ± 1.4 |
| Klinoptilolite | - | 5.4 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Götze, J.; Möckel, R.; Pan, Y. Mineralogy, Geochemistry and Genesis of Agate—A Review. Minerals 2020, 10, 1037. https://doi.org/10.3390/min10111037
Götze J, Möckel R, Pan Y. Mineralogy, Geochemistry and Genesis of Agate—A Review. Minerals. 2020; 10(11):1037. https://doi.org/10.3390/min10111037
Chicago/Turabian StyleGötze, Jens, Robert Möckel, and Yuanming Pan. 2020. "Mineralogy, Geochemistry and Genesis of Agate—A Review" Minerals 10, no. 11: 1037. https://doi.org/10.3390/min10111037
APA StyleGötze, J., Möckel, R., & Pan, Y. (2020). Mineralogy, Geochemistry and Genesis of Agate—A Review. Minerals, 10(11), 1037. https://doi.org/10.3390/min10111037