1. Introduction
Barite (BaSO
4) and celestite (SrSO
4) are the end-members of a nearly ideal solid solution. Despite its thermodynamics characteristics, this solid solution series shows a marked compositional bimodality [
1,
2] and samples with compositions that strongly differ from those of the two end-members are very rarely found in nature [
3]. Celestite (SrSO
4) is relatively common in a large diversity of sedimentary and igneous settings, where often appears scattered in small pockets [
4,
5]. Large deposits of celestite are rare and most of those that concentrate million metric tons appear associated with coastal marine carbonate and evaporite sequences [
6]. These exploitable deposits of celestite are thought to have formed during diagenesis due to the interaction of Sr-bearing aqueous solutions with sedimentary beds of calcium sulfate minerals, namely gypsum (CaSO
4·2H
2O) and/or anhydrite (CaSO
4) [
6]. This interaction would be the starting point for a dissolution-crystallization reaction. Thus, the sulfate released to the fluid phase upon dissolution of gypsum and/or anhydrite would react with the dissolved strontium and lead to the precipitation of celestite. Indeed, celestite pseudomorphs after gypsum microcrystals and cm-sized selenite crystals are abundant in the large celestite deposits of Granada (Spain) or Coahulia (Mexico), which gives support to the interpretation that relates the origin of these deposits to the development of interface coupled dissolution-precipitation (ICDP) reactions [
7]. Barite (BaSO
4) has a much broader geological distribution than celestite ([
3] and references therein). Most barite deposits form through the mixing of sulfate-rich fluids, such as seawater, with fluids charged with Ba after their interaction with silicate minerals [
3]. Although barite can also form after gypsum and/or anhydrite [
4,
8], barite deposits associated to sulfate evaporite sequences are volumetrically much less relevant than their celestite counterparts [
5,
6].
ICDP reactions play a major role in the re-equilibration between mineral phases and aqueous fluids in the Earth’s crust. Evidences of the development of these reactions are found in a plethora of geological settings, including sedimentary, diagenetic, metamorphic, and metasomatic environments [
9,
10]. ICDP reactions initiate with the formation of nuclei of a secondary phase on the surface of a primary one. These reactions then proceed through the inwards advance of a sharp reaction front located at the interface between a progressively thicker rim of the secondary phase and a shrinking core of the primary one. This inwards advance of the reaction front can only take place if communication between the fluid phase and the reaction front is continuously maintained. In those ICDP reactions that involve a negative molar volume change this communication is guaranteed by a network of interconnected porosity that develops concomitantly to the formation of the secondary phase [
11]. This porosity balances the volume reduction associated to the reaction and thereby allows the preservation of the external shape of the primary phase [
9,
12,
13,
14]. This explains that most ICDP reactions are pseudomorphic [
9,
12,
13,
14]. The volume of porosity generated during ICDP reactions is not exclusively defined by the difference in molar volume between the phases involved. A further porosity contribution arises when the secondary phase is less soluble than the primary one [
14,
15,
16]. Finally, factors like the solid to fluid volume ratio [
17], the evolution of the composition of the fluid at the reaction front [
18], the existence/absence of crystallographic relationships between the phases involved in the ICDP reaction [
19], the presence of pre-existing cracks or other defects within the primary phase crystals [
20] as well as the specific morphological and textural features of the secondary phase [
21,
22,
23,
24,
25,
26] can also contribute to modulate the volume of porosity generated during ICDP reactions and, more importantly, to define the spatial arrangement of the ICDP reaction-related porosity within the secondary phase [
27,
28,
29]. The permeability and tortuosity of the thus-generated porosity network effectively define the degree of communication between the fluid phase and the reaction front. It has, as a result, a strong impact in the kinetics of the ICDP reactions.
In this work, we present an experimental study of the interaction of gypsum fragments with Ba and Sr-bearing solutions. Since barite and celestite have smaller molar volumes and solubilities than those of gypsum, it can be expected that this interaction will result in the pseudomorphic replacement of the gypsum crystals by the former phases through the development of ICDP reactions. By comparing the characteristics of these two mineral replacement processes, we aim to better understand how the kinetics of ICDP reactions is influenced by intrinsic (differences in molar volume, solubility, crystallographic similarities between the involved phases) and extrinsic (composition of the fluid phase) factors.
4. Discussion
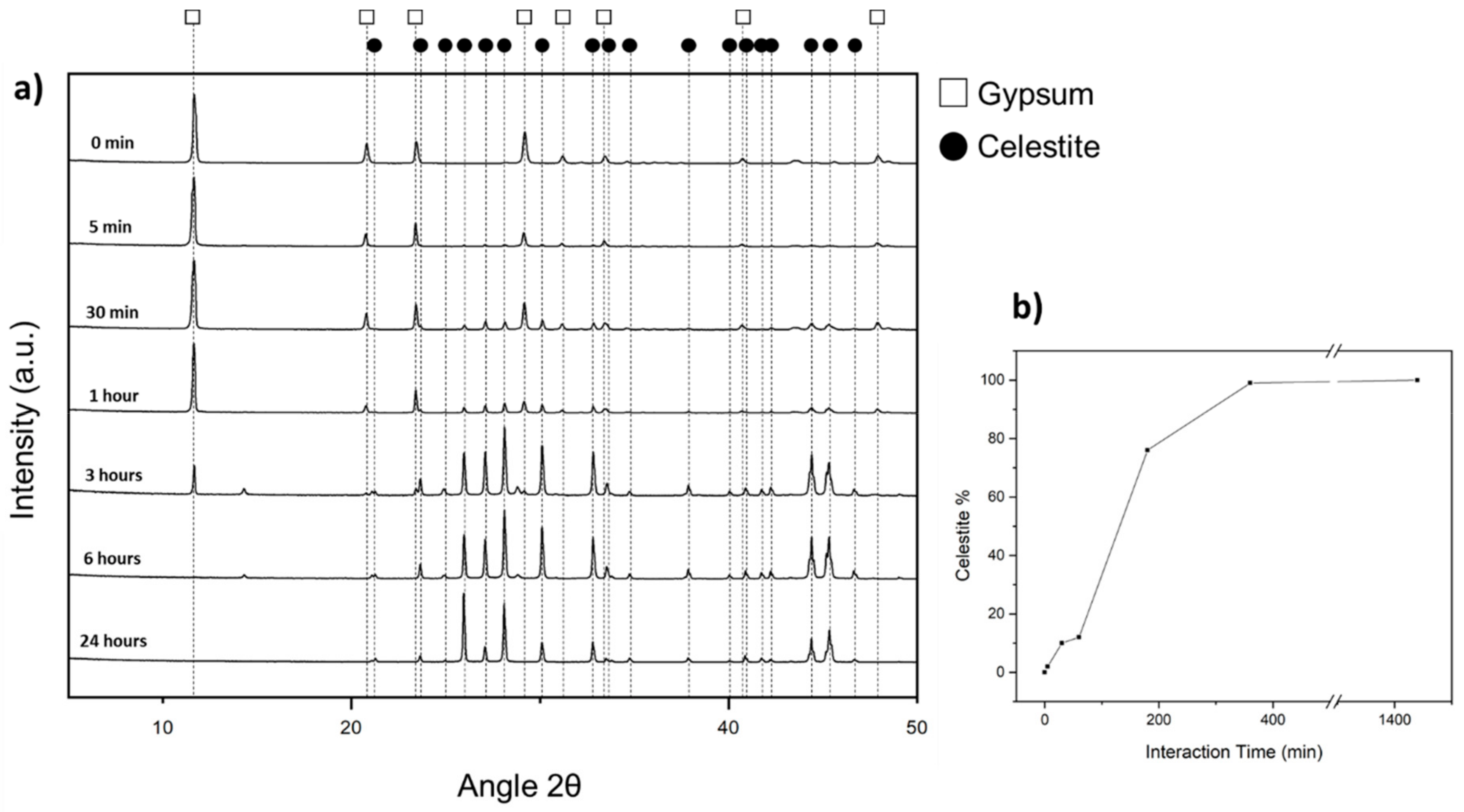
Upon interaction with Sr and Ba-bearing aqueous solutions, gypsum crystals transform into celestite and barite, respectively. In both cases, the transformation initiates at the surface of the gypsum crystal and advances inwards, defining a sharp reaction front that separates a polycrystalline transformed layer from the remaining unreacted core of gypsum. Furthermore, the original external shape of the gypsum crystal is preserved throughout the transformation process. These transformation features are consistent with the transformation taking place through an ICDP reaction. The development of these type of reactions involves the definition of a dissolution-crystallization feedback loop [
9,
12]. This loop guarantees the coupling through the interface of the rates of the primary phase dissolution and the secondary phase precipitation. In the cases of the transformation of gypsum into celestite and into barite, this feedback loop arises as, upon interaction with the aqueous solution, gypsum starts to dissolve and releases SO
42− and Ca
2+ ions to the liquid phase. The aqueous solution then becomes supersaturated with respect to celestite, if it bears Sr
2+, or barite, if it bears Ba
2+, which eventually leads to the precipitation of these phases. The depletion of the aqueous solution in SO
42− that results from this precipitation in turn further promotes the dissolution of gypsum and, consequently, the release of SO
42− and Ca
2+ ions to the liquid phase, thereby feeding the dissolution-crystallization loop. This loop should effectively operate until the complete replacement of the gypsum grains by celestite (or barite) provided that throughout the whole process the concentrations of dissolved Sr
2+ (or Ba
2+) ions at the gypsum-celestite (or barite) interface are high enough to allow the precipitation of celestite (or barite) through reaction with the released to the liquid SO
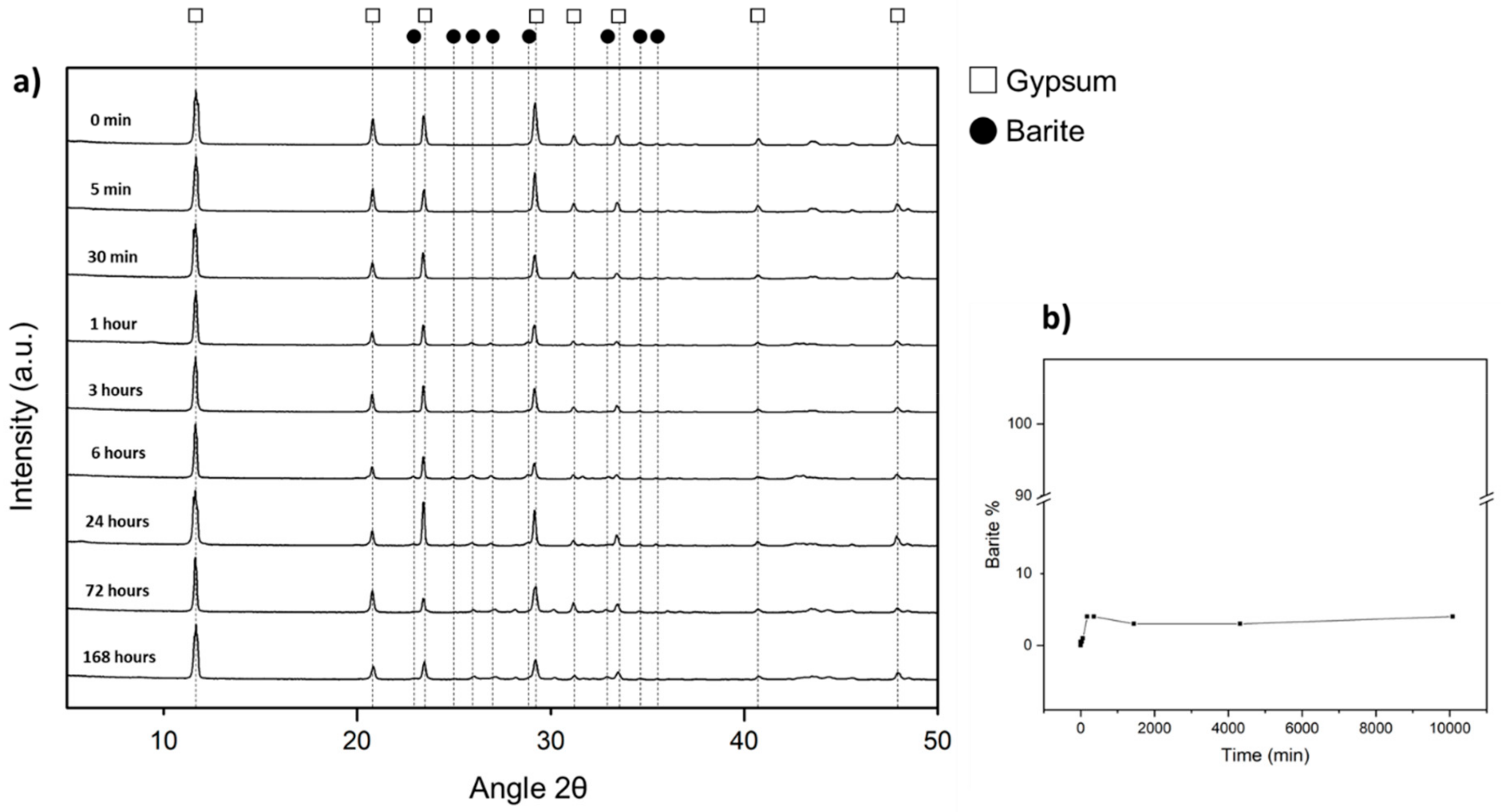
42− ions. Our results show that, indeed, this is the case when a gypsum crystal reacts with an aqueous solution that bears 0.5 M Sr. We observe that in less than 24 h this interaction results in the pseudomorphic replacement of the whole gypsum crystal by an aggregate of celestite crystals. In contrast, when the gypsum crystal interacts with a 0.5 M Ba-bearing aqueous solution, the gypsum dissolution-barite precipitation loop only works efficiently during a very short time, leading to the formation of a less than 30 µm thick layer of barite crystals that surrounds an unreacted gypsum core during the first hour of the interaction. Afterwards, no further advance of the gypsum into barite transformation is observed.
As explained in the introduction section, the starting point of an ICDP reaction is the nucleation of the secondary phase on the surface of the primary one. Henceforth, for the reaction to progress the continuous communication between the primary phase-secondary phase interface and the bulk solution is required [
16]. This communication is guaranteed by the generation of porosity concomitantly to the progress of the transformation when the ICDP reaction involves a negative molar volume change [
9,
12]. More porosity can be added when the ICDP reaction also involves a secondary phase that is less soluble than the primary one [
14,
15,
16]. Both criteria for the generation of porosity are fulfilled in the cases of the pseudomorphic replacement of gypsum by celestite and by barite. Indeed, both celestite and barite have molar volumes that are smaller than the molar volume of gypsum (V
Clt = 46.25 cm
3/mol, V
Brt = 52.11 cm
3/mol, V
Gp = 74.31 cm
3/mol). Similarly, celestite and barite are less soluble than gypsum (K
spClt = 10
−6.63; K
spBrt = 10
−9.97; K
spGp = 10
−4.58; PHREEQC database). Consequently, at any time during the pseudomorphic replacement of gypsum by celestite or barite the transformed layer that surrounds the unreacted gypsum core contains a volume of porosity that is equal (higher if the solubility-related contribution is taken into account) to the volume loss derived from the molar volume change associated to the replacement reaction (ΔV = −27.06 cm
3/mol and −22.20 cm
3/mol for the transformation into celestite and barite, respectively): i.e., ≥36.4% porosity when gypsum transforms into celestite and ≥30.3% porosity when it transforms into barite.
The large volume of porosity generated during the transformation of gypsum into celestite facilitates the continuous communication between the gypsum-celestite interface and the bulk solution along the replacement process, explaining the rapid kinetics of the ICDP reaction responsible for this replacement process. This communication is facilitated by the spatial arrangement of this porosity, which appears structured within celestite pseudomorphs from surface to core: (i) in open spaces between randomly oriented celestite crystals, in Region 1, (ii) in channels between approximately parallel elongated celestite crystals, in Region 2, and finally (iii) in large open spaces between celestite spherulitic aggregates, in Region 3, and randomly oriented elongated celestite crystals, in Region 4 (
Figure 5). This organization results in a network of interconnected pores that facilitates the transfer of Sr
2+ ions from the bulk solution to the interface, where they react with the SO
42− ions released during gypsum dissolution to precipitate secondary celestite.
The specific features of porosity organization within celestite pseudomorphs after gypsum are the consequence of the combined influence of two factors: (i) celestite crystals nucleate randomly oriented on gypsum surface at the very early stages of the replacement process, and (ii) the release of SO
42− ions during gypsum dissolution is accompanied by the release of an equal amount of Ca
2+ ions and double amount of water molecules. The first factor determines the textural organization of Region 1 in celestite pseudomorphs after gypsum. Furthermore, this factor is also in the origin of the textural organization of Region 2, whose stockade-like structure (
Figure 3a,d;
Figure 4a,c) develops as differently oriented celestite crystals in Region 1 compete for space during growth. Celestite growth is faster along [001] direction. Those crystals in Region 1 that are oriented so that their [001] direction is perpendicular to the original gypsum surface are in advantage and can grow without interference. In contrast, the development of those crystals differently oriented will be prevented by the lack of space. This competitive growth mechanism rapidly leads to the formation of the parallel column-like celestite crystals that constitute Region 2 of celestite pseudomorphs after gypsum. Competitive growth is a common phenomenon during mineral replacement processes that can significantly influence the texture of the resulting pseudomorph when the secondary phase has a preferential growth direction. Obviously, the randomly oriented nucleation of the secondary phase on the surface of the primary one is a prerequisite for the occurrence of competitive growth phenomena. Stockade-like structures that have been attributed to competitive growth have been described in pseudomorphs of calcite after gypsum [
27] and metallic gold after calaverite (AuTe
2) [
32]. In both cases, the early nucleation of the secondary phase on the surface of the primary one was randomly oriented. The transition from parallel column-like celestite crystals in Region 2 to isolated spherulitic aggregates in Region 3 in celestite pseudomorphs after gypsum can be the consequence of the progressive development of a gap between the transformed layer and the unreacted gypsum core. This gap, which is clearly visible in
Figure 3 and
Figure 4, forms due to the difficulty to accommodate within the stockade-like structure the porosity volume required to balance the very large molar volume change associated to the gypsum into celestite transformation. The formation of similar gaps has been observed in other mineral replacement processes involving large negative molar volume changes, like the replacements of gypsum by calcite [
27] and pyrochlore by rutile/anatase [
33]. The composition of the aqueous solution that fills this gap will evolve as the gypsum into celestite transformation progresses. The concentration of Sr
2+ in this solution will be the result from the balance between the amount of Sr
2+ transferred from the bulk solution and that consumed through the precipitation of celestite. This concentration will decrease as the transformation progresses. The diluting effect derived from gypsum dissolution, which together to SO
42− and Ca
2+ ions also releases water molecules to the aqueous solution that fills the gap, will further contribute to reduce Sr
2+ concentration. Simultaneously, the Ca
2+/Sr
2+ concentration ratio will increase. The growth of celestite in a progressively Ca-richer environment could result in the incorporation of a small amount of Ca
2+ substituting Sr
2+ into celestite structure. This incorporation is consistent with EDX analyses of spherulites, sheaf like and column-like celestite crystals. Since the ionic radii of these cations (ratio Ca
XII = 1.34; ratio Sr
XII = 1.44 Å) is significantly different, this incorporation would introduce lattice strain into celestite structure. This strain could then be released through the formation of dislocations. The distribution of these dislocations in regularly spaced arrays in small angle grain boundaries can explain the progressive loss of parallelism between column-like celestite crystals in Region 2 the farer they are from the surface of the celestite pseudomorph. As the Ca
2+/Sr
2+ concentration ratio in the interface solutions continuous to grow due to both, Sr
2+ consumption through celestite growth and Ca
2+ release through gypsum dissolution, the growth of column-like celestite crystals can become inhibited. Celestite crystallization will then only resume after the supersaturation required for the formation of new celestite nuclei is overcome thanks to the transfer of Sr
2+ ions from the bulk solution to the interface. Since these nuclei grow in a Ca
2+-rich environment, it is to be expected that they incorporate small amounts of Ca. Sánchez-Pastor et al. [
34] demonstrated that celestite crystal habit is very sensitive to isomorphic substitutions. Again, the relaxation of Ca incorporation-related lattice strain through the formation of dislocations can explain that these nuclei grow to form the sheaf-like and spherulitic celestite aggregates that constitute the Region 3 of celestite pseudomorphs. EDX analysis of these aggregates confirm that they contain small amounts of Ca. Maintaining the aqueous solution at the interface supersaturated with respect to celestite becomes progressively more difficult as the transformed layer that surrounds the unreacted gypsum core is thicker and the transfer of Sr
2+ ions from the bulk solution requires a longer time. Growth under lower supersaturation could explain the celestite morphological transition from sheaf-like and spherulitic aggregates to small elongated crystals that marks the boundary between Regions 3 and 4 in celestite pseudomorphs after gypsum. It has been demonstrated that high supersaturation kinetically promotes isomorphic ion incorporation above the thermodynamic equilibrium [
35]. It is worthwhile to note that the Ca content of celestite crystals in the latter region is below the EDX analysis detection limit, this is, they are basically pure celestite and their habit show characteristics that resemble those of pure celestite crystals grown in a system where mass transfer is mainly control by diffusion [
34].
Though smaller than the molar volume change involved in the transformation of gypsum into celestite, a large negative molar volume change is also involved in the transformation of gypsum into barite. This means that the pseudomorphic replacement of gypsum by barite must be accompanied by the generation of a large volume of porosity. This porosity should facilitate the progress of this replacement at a rate similar to that of the replacement of gypsum by celestite. Surprisingly, this is not so. The facts that (i) the interaction of gypsum crystals with 0.5 M Ba-bearing aqueous solutions only leads to the replacement by barite on a thin most external layer and, (ii) this layer forms shortly after the beginning of the interaction, indicate that the gypsum crystal surface rapidly becomes passivated. This passivation prevents that the gypsum dissolution-barite precipitation feedback loop can effectively operate. Based on SEM imaging observations and 2D-XRD analysis results Ruiz-Agudo et al. [
36] recently attributed this passivation to the formation of a thin layer of epitactic barite on the gypsum surface. Indeed, there are numerous examples of mineral surface passivation by epitactic overgrowths of a secondary phase [
18,
26,
37,
38]. However, this passivation commonly is only partial and only leads to the slowdown of the ICDP reaction. This occurs, for example, during the interaction between anhydrite and a Ca-bearing aqueous solution [
21]. Although calcite (CaCO
3) crystals grow oriented on anhydrite surfaces, the existence of epitactic relationships between both phases do not prevent the formation of fully transformed pseudomorphs of calcite after anhydrite [
21]. A similar behaviour is observed during the interaction of anhydrite with Pb-bearing aqueous solutions, which leads to the formation of epitactic anglesite (PbSO
4) layers on the surface of anhydrite [
24]. In these two examples the epitaxy involves phases that are not isostructural but share some common structural features. The misfit through the interface between the structures of the primary and secondary phases determines that the epitactic growth on the latter on the former takes place through a Volmer–Weber mechanism [
39,
40]. This mechanism involves the formation of oriented 3D nuclei that grow to coalesce and form an epitactic layer. This layer can completely carpet the surface of the substrate. However, because the substrate commonly contains symmetry operators, there are several equally probable symmetry related orientations for the epitactic 3D nuclei. When differently oriented 3D nuclei coalesce, they leave micropores between crystal grains. Consequently, the so-formed epitactic layers contain a certain amount of microporosity and are unable to completely armour the substrate from interaction with an aqueous solution [
21,
24,
26]. Only when the primary and secondary phases involved in an ICDP reaction are isostructural and have similar lattice parameters the formation of an epitactic overgrowth can lead to the complete stoppage of the ICDP reaction. This has been observed, for example, during the interaction of calcite {10
14} surfaces with Cd-bearing aqueous solutions [
37]. In this case, this interaction leads to the rapid formation of a nanometric epitactic layer of isostructural otavite (CdCO
3) on the calcite surface. The almost perfect match between the structures of calcite and otavite through the interface facilitates that the epitactic growth takes place through a Frank van der Merwe or a Stranski-Krastanov epitactic mechanism [
41,
42,
43]. These mechanisms involve the growth and coalescence of epitactic 2D nuclei and the perfect substrate armouring that the so-formed epitactic layer explains the extremely low ability of calcite surfaces to remove Cd
2+ ions form aqueous solutions through coprecipitation processes.
Barite and gypsum are not isostructural. Furthermore, the epitactic relationships between barite and gypsum reported by Ruiz-Agudo et al. [
36] involve the parallelism of (010)
Gp and {001}
Brt planes and only one crystallographic direction contained within these planes ([100]Gp ‖ [010]Brt). Despite the low mismatch (−3.8%) between [100]Gp and [010]Brt, it is very unlikely that a complete substrate armouring could result from the formation of an uni-directional epitactic layer. Interestingly, despite celestite and anglesite being isostructural with gypsum, these phases grow randomly oriented on gypsum (010) substrates [
39,
44]. Moreover, as explained in the Results section, our SEM observations do not clearly support an epitactic growth of barite on gypsum surfaces. Indeed, most barite crystals (
Figure 7) appear with their large face (001) laying approximately parallel to the gypsum (010) form. However, numerous barite crystals show other orientations. More importantly, not all barite crystals that lay with their (001) plane parallel to (010)
Gp are equally oriented but many appear differently rotated with respect to their [001] direction. This crystal arrangement could be the result of barite crystals forming in the layer of aqueous solution closest to the gypsum (010) surface through a homogeneous nucleation mechanism and the settling by gravity on the gypsum surface. Because these barite crystals are extremely platy, this settling mechanism would lead to most of them laying with their most developed face parallel to the gypsum substrate surface. Furthermore, this settling mechanism would also account for the dispersion in the rotation of these crystals around their [001] axis. It is important to take into account that barite is very sparingly soluble and as soon as gypsum starts to dissolve in contact with a 0.5 M Ba-bearing aqueous solution, the supersaturation with respect to barite of the solution layer immediately in contact with the gypsum surface will be extremely high. This can make it possible that the barrier for barite homogeneous nucleation is overcome and barite nuclei form in this solution layer. It is also worthwhile to note that the morphology of the barite crystals that constitute the transformed rim around the unreacted gypsum crystal closely resemble the habit of barite crystals that precipitate by rapidly mixing aqueous solutions [
45]. In such a system barite nucleation is homogeneous and takes place under very high supersaturation conditions. The proposed crystal settling mechanism would lead to the formation of a transformed layer that could contain a certain degree of porosity. However, a large porosity volume alone does not guarantee an effective pathway for mass transfer from the bulk solution to the primary phase-secondary phase interface. Interconnection and tortuosity are main properties which play a more important role than porosity volume in defining mass transfer rates through porosity networks. The interconnection of a porous network that results from the parallel stacking of platy crystals will be low while its tortuosity will be high. Consequently, regardless of what the origin of the arrangement of barite crystals in the transformed layer around the unreacted gypsum crystal could be, epitactic growth or settling by gravity, the morphology of these barite crystals arises as a critical factor that contributes to completely armor the gypsum substrate and leads to the stoppage of the interface coupled gypsum dissolution-barite crystallization reaction.
It is well known that most of the largest deposits of celestite in the world are associated to the evaporate-carbonate environment realm [
3]. Although a genetic relationship between these deposits and massive precipitation during the early stages of evaporation of seawater was early proposed [
46,
47], this hypothesis was soon discarded since it was not supported by experimental or field observations [
3]. Most authors support hypotheses that connect the formation of celestite massive deposits to processes that take place during burial diagenesis [
3,
6,
48,
49]. These processes would involve the interaction of carbonates and calcium sulfates with basinal fluids that have anomaly high Sr/Ba ratios after undergoing Sr enrichment due to (i) the diagenetic alteration of Sr-rich carbonates, that releases Sr to the basinal fluid and (ii) continuous barite precipitation, which removes Ba from the basinal fluid [
3,
6]. The results of our study support that the interaction of a calcium sulfate phase, like gypsum, with a Ba-bearing aqueous solution results in the rapid precipitation of barite. However, it is unlikely that this interaction can lead to the formation of barite massive deposits through the replacement of calcium sulfates, as gypsum surfaces become rapidly passivated due to barite precipitation. Due to the large difference between the solubility products of the endmembers of the barite-celestite solid solution, precipitates that result from the interaction of fluids with an extremely wide range of Ba/Sr ratios with calcium sulfate evaporites will be almost pure barite [
3,
6,
50,
51]. Consequently, this interaction is likely to be a mechanism for the progressive Sr-enrichment of basinal fluids to Sr/Ba ratios that allow for the formation of celestite. Our results demonstrate that, if gypsum crystals are in contact with a Sr-rich fluid, they can be rapidly replaced by celestite through an ICDP reaction. The fast kinetics of this replacement reaction and the fact it results in local rather that disperse precipitation of celestite gives credit to the replacement of calcium sulfates by celestite hypothesis to explain the genesis of massive celestite deposits in sedimentary basins.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








