
The Timing and Effects of Low-Dose Ethanol Treatment on Acetaminophen-Induced Liver Injury

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Model and Treatments
2.3. Measurement of Alanine Transaminase Levels in Serum
2.4. Measurement of GSH Levels in the Liver
2.5. Histology and Immunohistochemistry
2.6. Measurement of TNF-α and IL-6 in the Liver
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
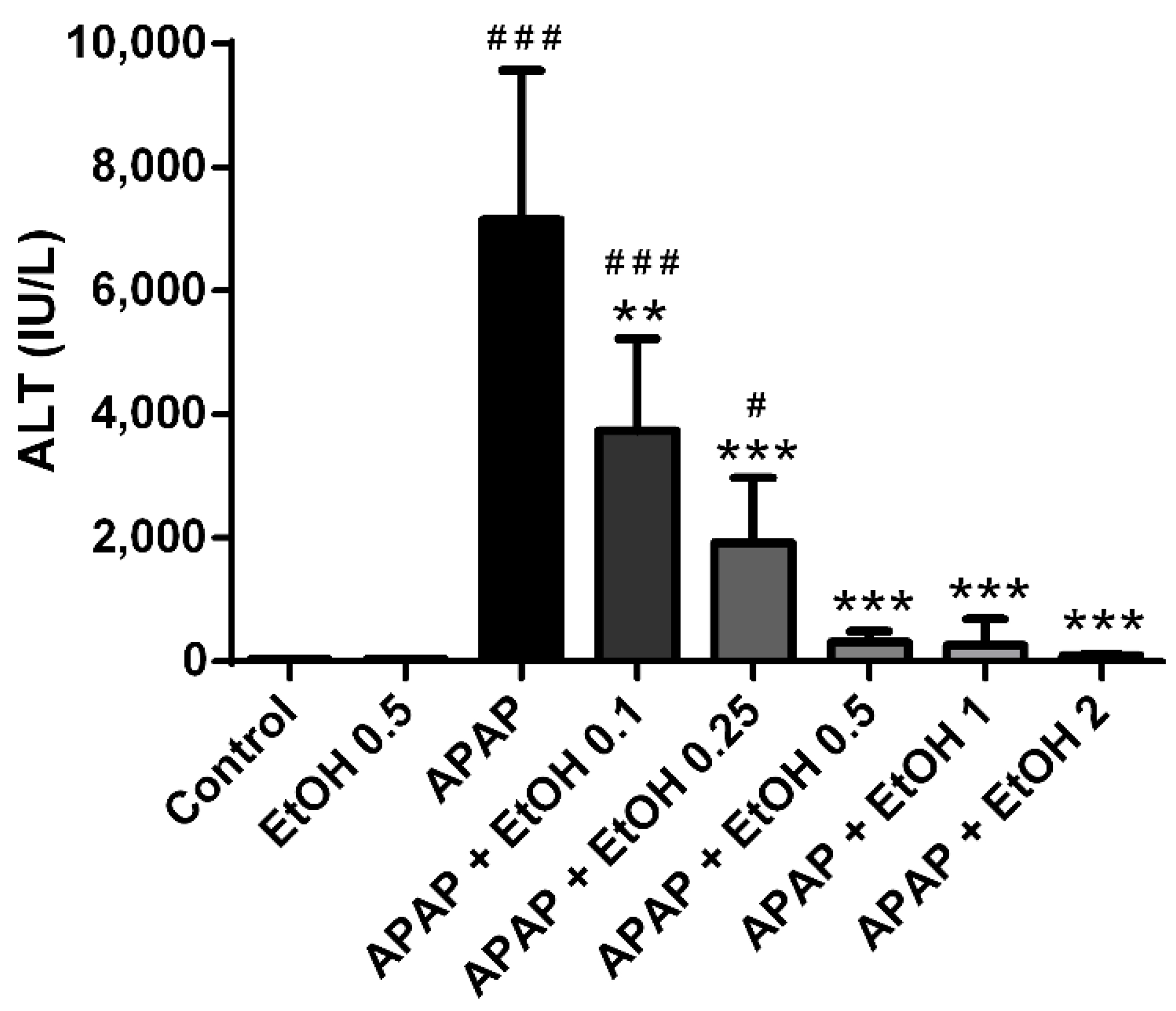
3.1. Effects of Different Doses of Ethanol on Serum ALT Levels in APAP-Induced Liver Injury
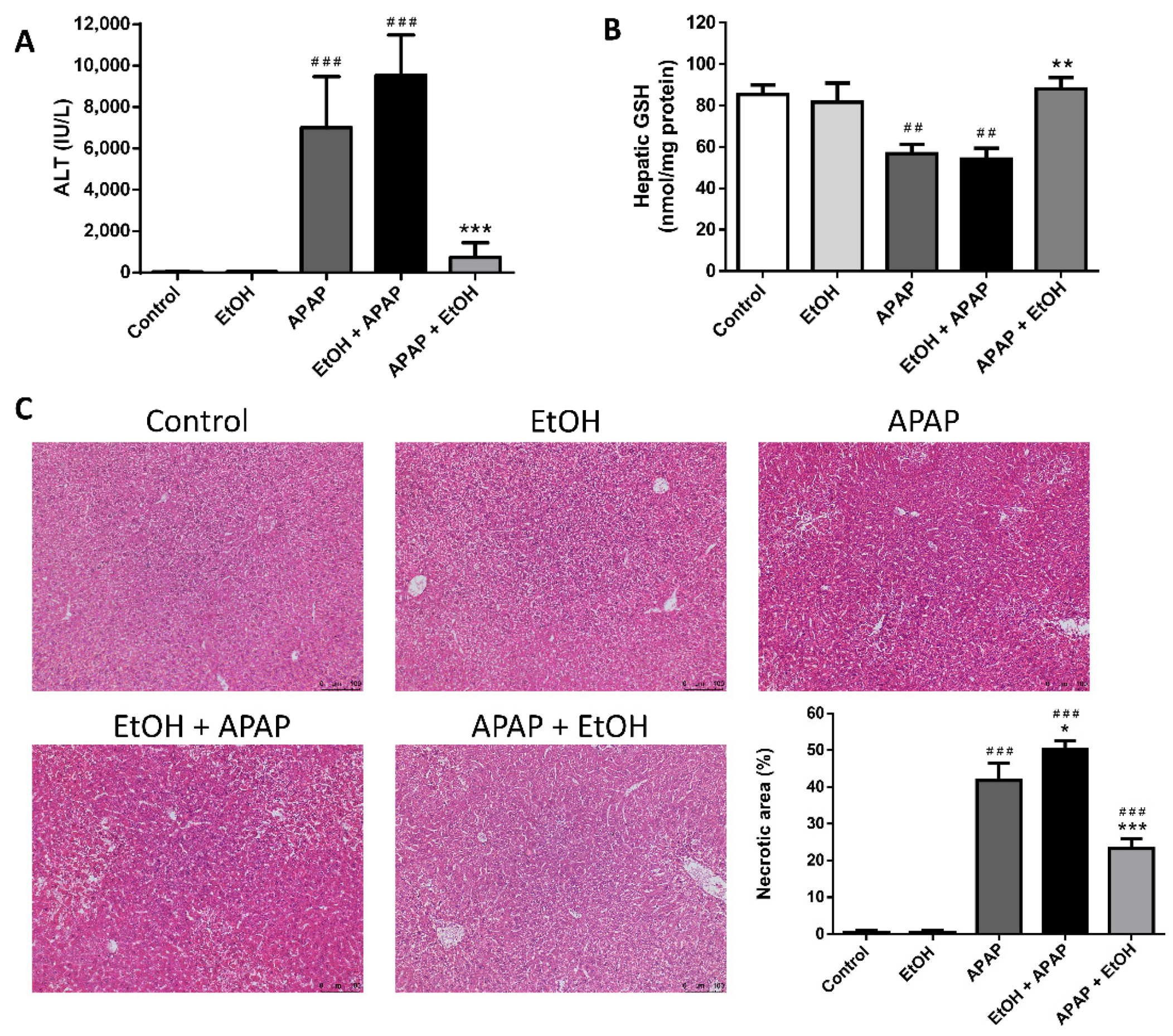
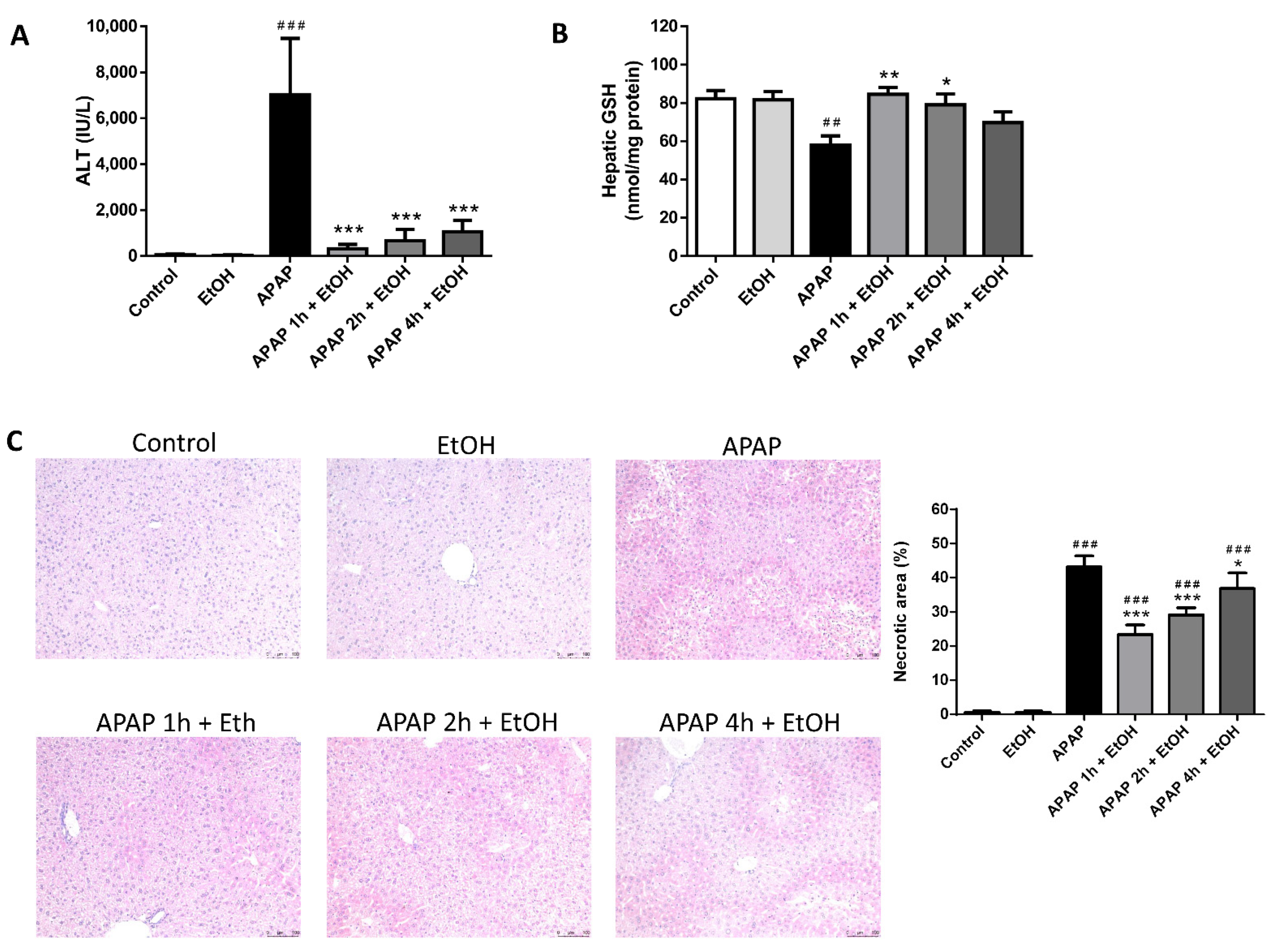
3.2. Effect of Ethanol Pre- and Post-Treatment on Serum ALT Levels, Hepatic GSH Levels, and Hepatic Histological Changes
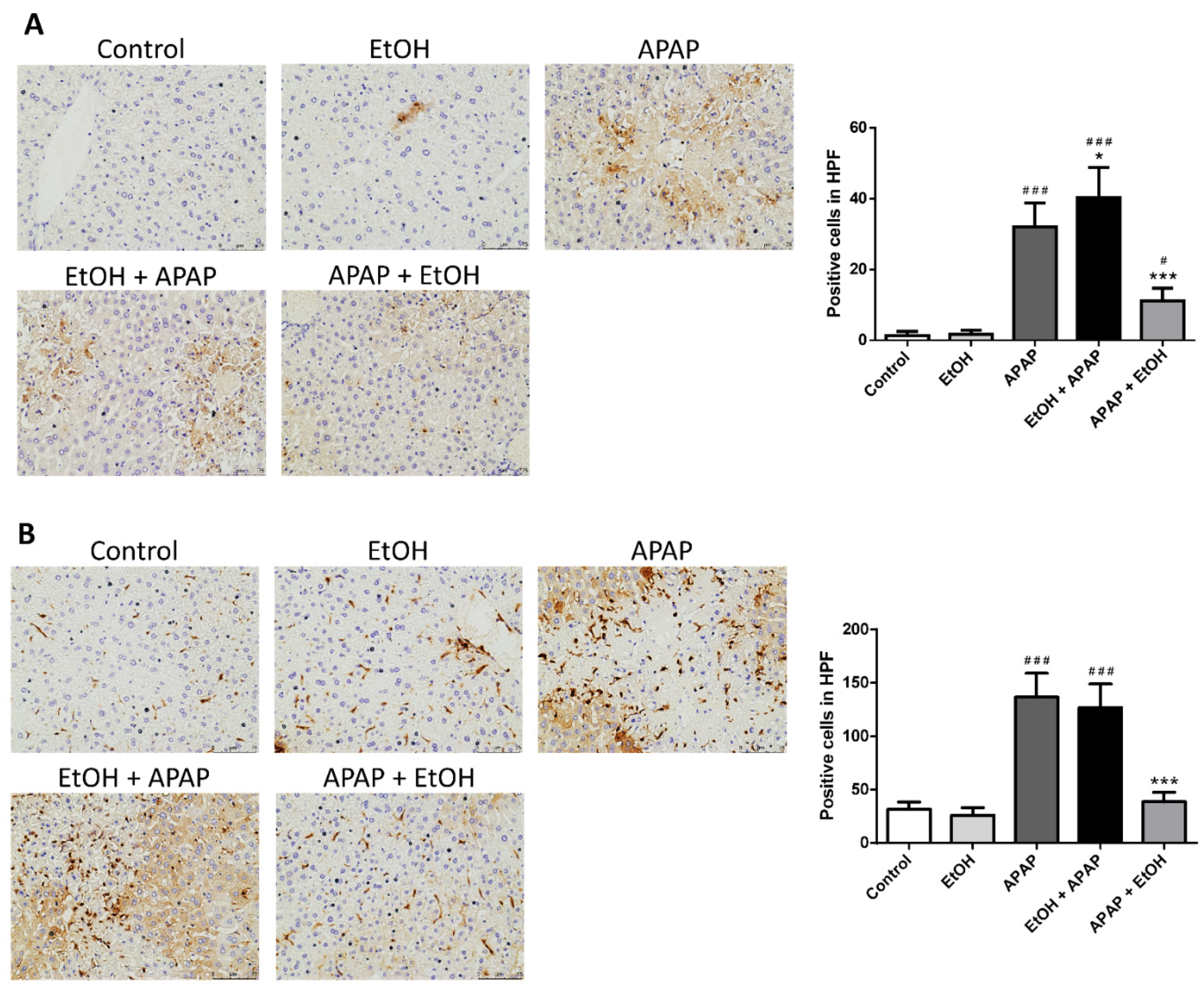
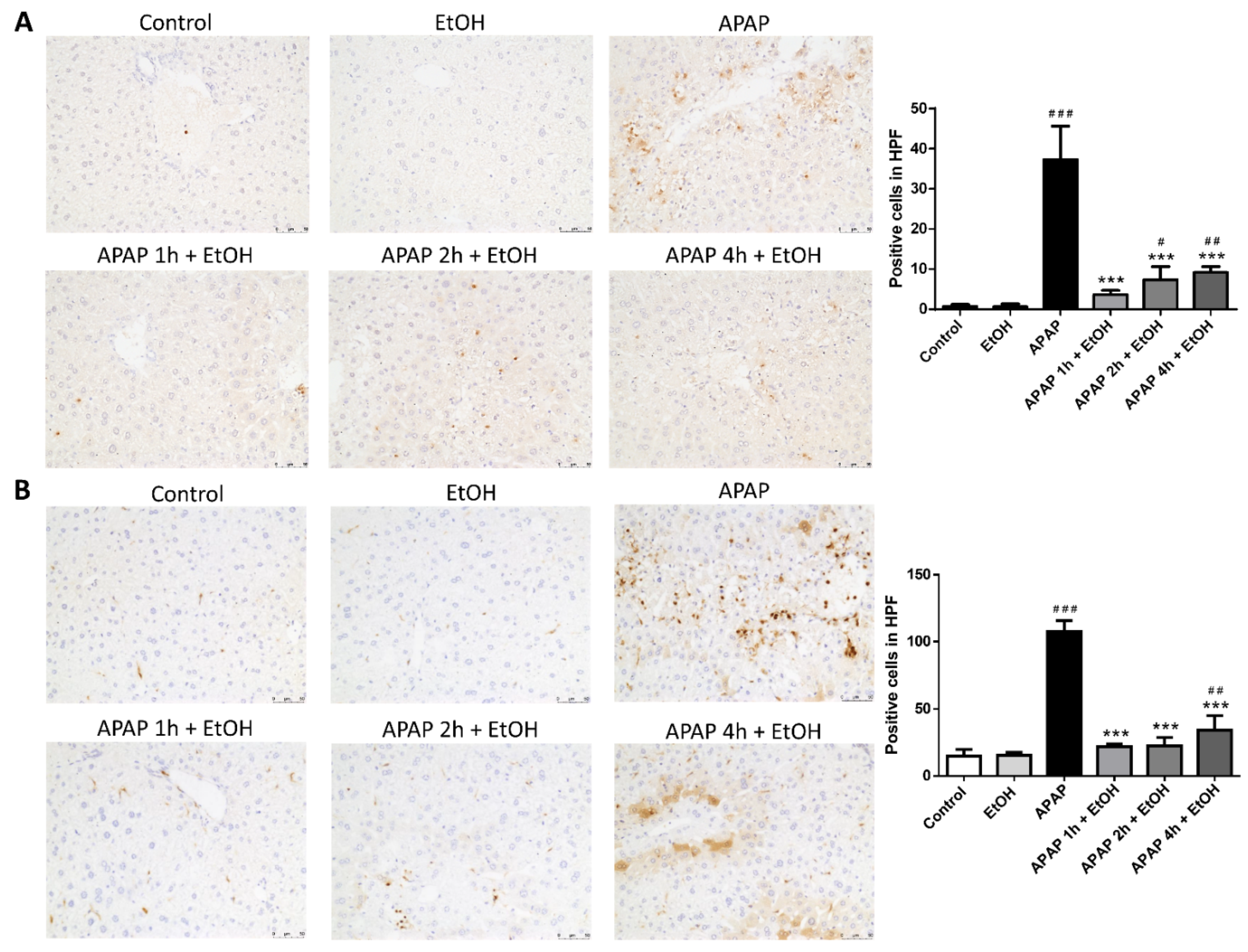
3.3. Effects of Ethanol Pretreatment and Posttreatment on the Infiltration of Neutrophil and Macrophage in the Liver
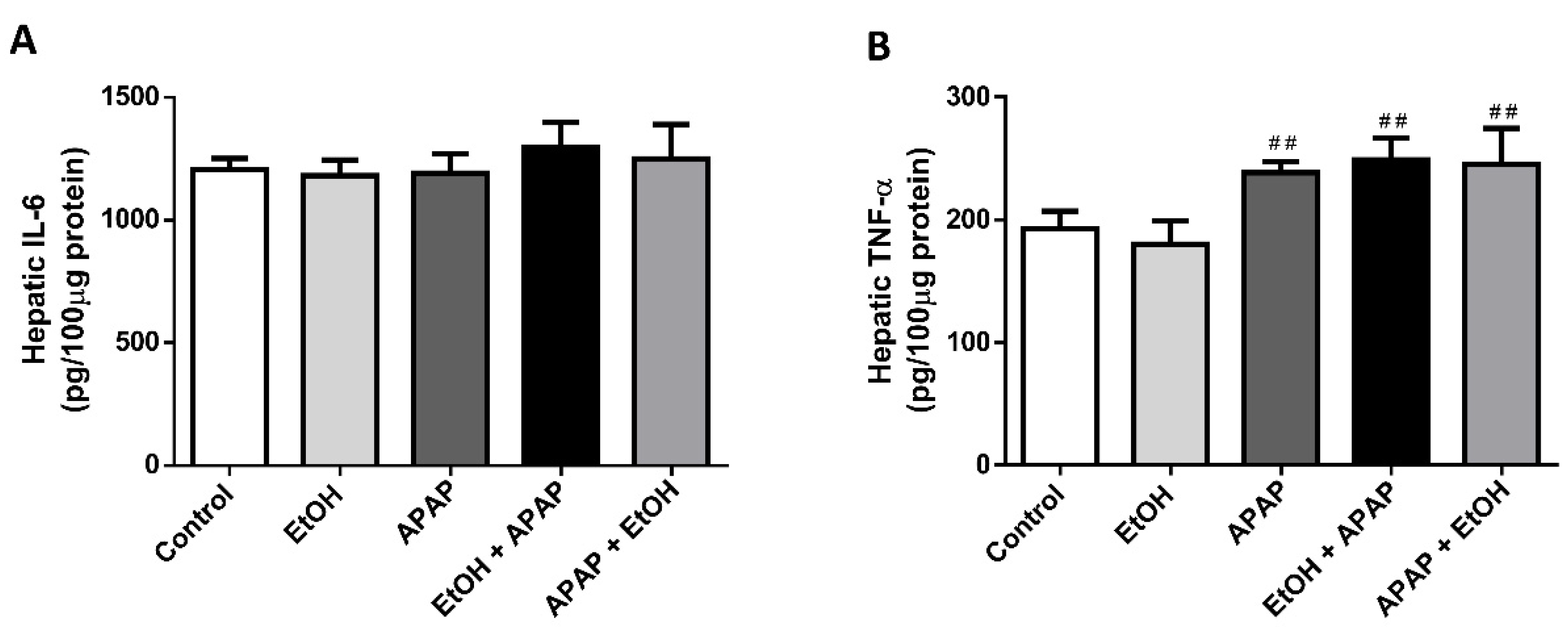
3.4. Effects of Ethanol Pre- and Post-Treatment on Hepatic TNF-α and IL-6 Levels
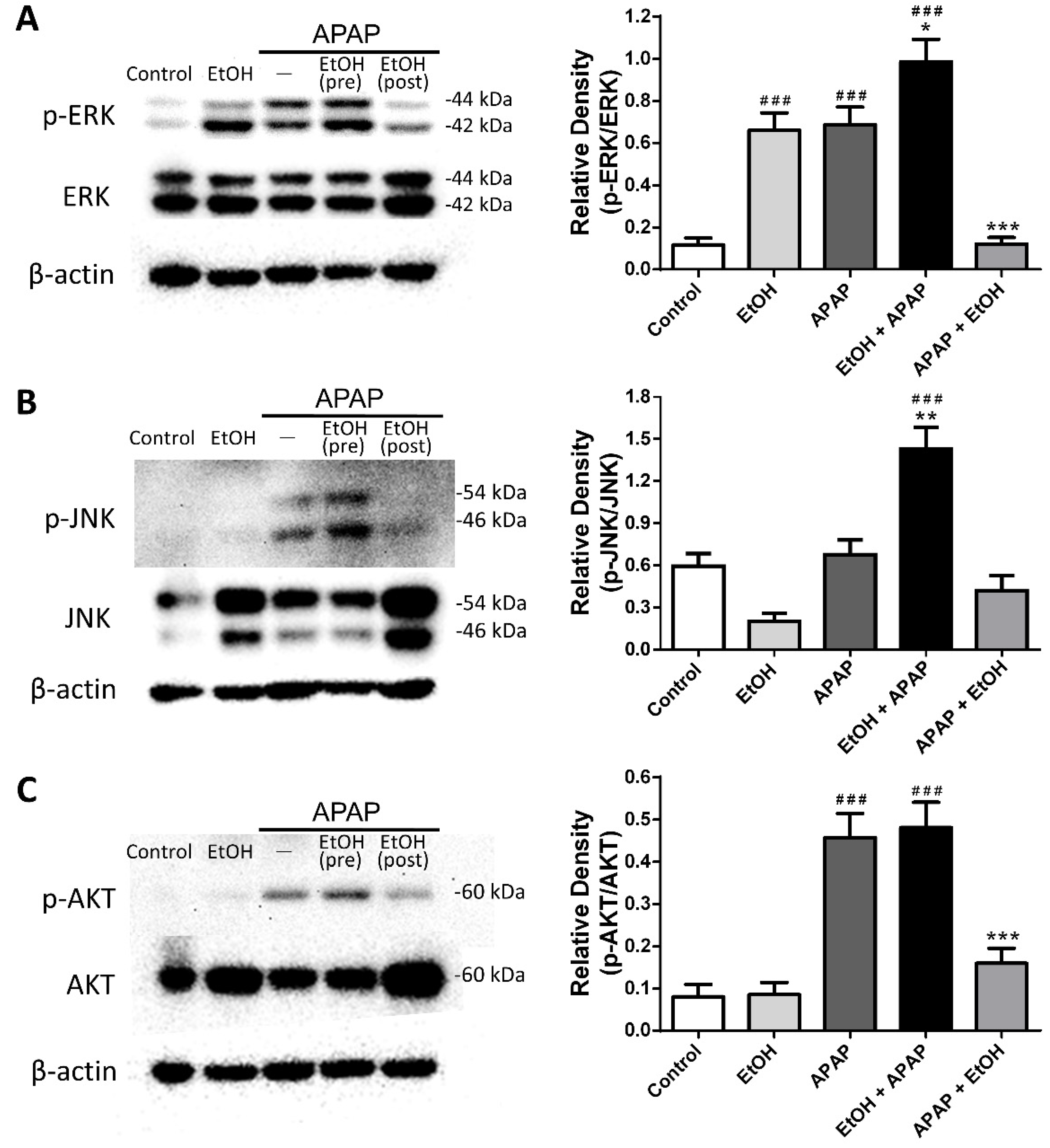
3.5. Effects of Ethanol Pre- and Post-Treatment on ERK, JNK, and AKT Expression and Phosphorylation in the Liver
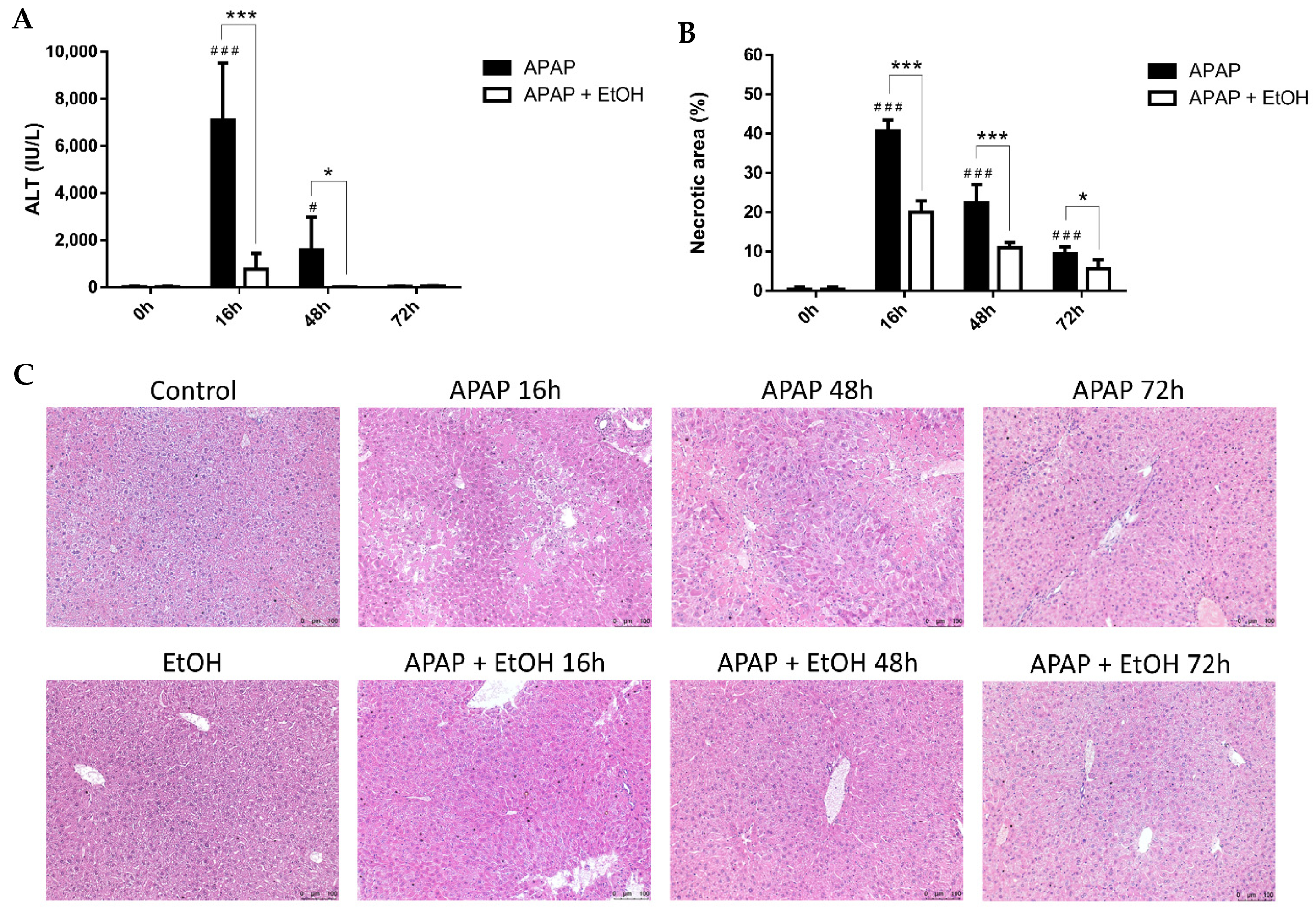
3.6. Effects of Different Schedules of Ethanol Treatment on APAP-Induced Liver Injury
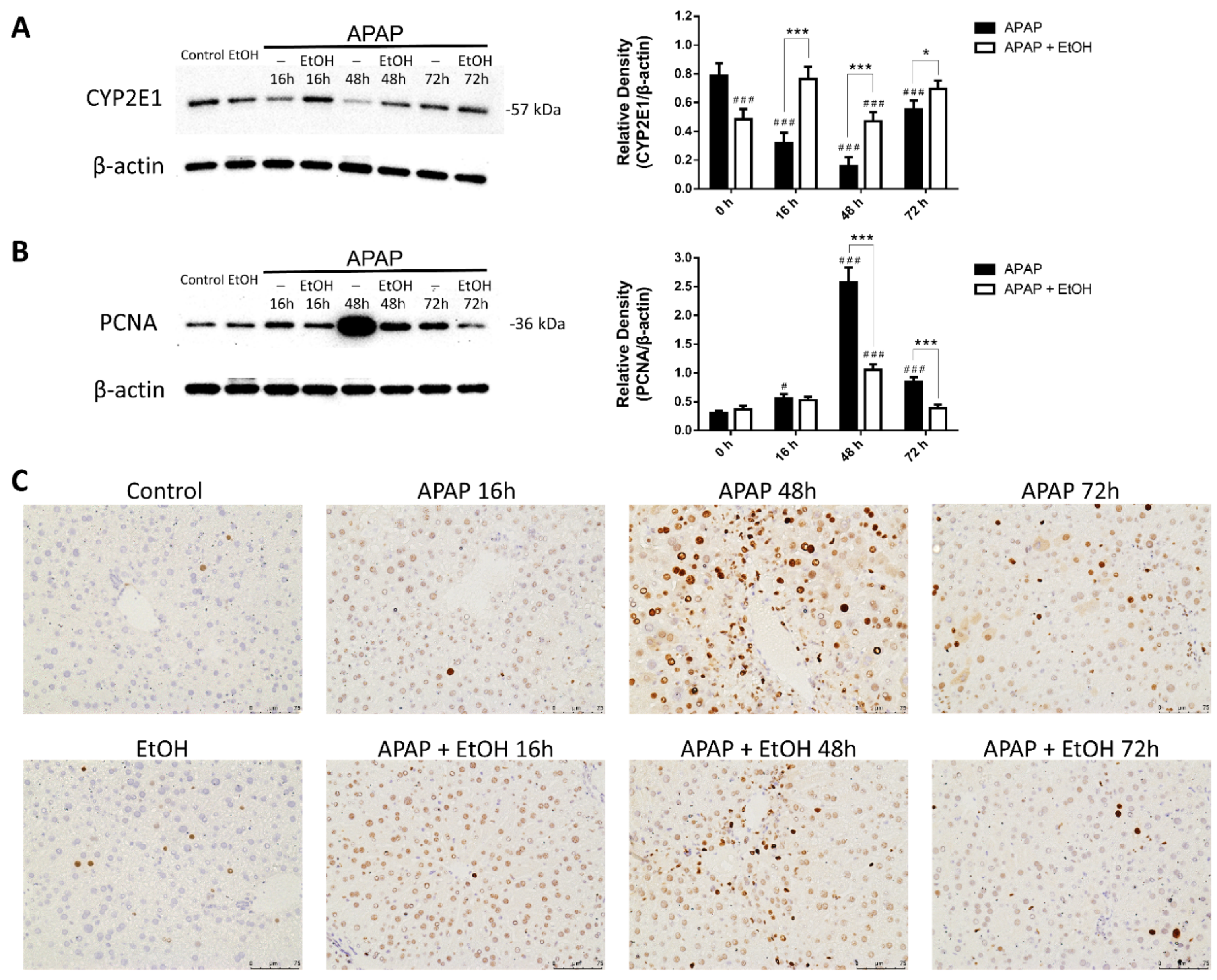
3.7. Effects of Ethanol Treatment on Liver Regeneration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bunchorntavakul, C.; Reddy, K.R. Acetaminophen-related hepatotoxicity. Clin. Liver Dis. 2013, 17, 587–607. [Google Scholar] [CrossRef]
- Murray, K.F.; Hadzic, N.; Wirth, S.; Bassett, M.; Kelly, D. Drug-related hepatotoxicity and acute liver failure. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.M.; Hiatt, J.R.; Zarrinpar, A. Acetaminophen hepatotoxicity: An updated review. Arch. Toxicol. 2015, 89, 193–199. [Google Scholar] [CrossRef]
- Schilling, A.; Corey, R.; Leonard, M.; Eghtesad, B. Acetaminophen: Old drug, new warnings. Cleve Clin. J. Med. 2010, 77, 19–27. [Google Scholar] [CrossRef]
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548. [Google Scholar] [CrossRef]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.R.; Sharpe, M.R.; Williams, C.D.; Taha, M.; Curry, S.C.; Jaeschke, H. The mechanism underlying acetaminophen-induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J. Clin. Investig. 2012, 122, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Briedé, J.J.; Jennen, D.G.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.; de Kok, T.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [Green Version]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol. Res. 2013, 35, 6–16. [Google Scholar] [PubMed]
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [Green Version]
- Quertemont, E. Genetic polymorphism in ethanol metabolism: Acetaldehyde contribution to alcohol abuse and alcoholism. Mol. Psychiatry 2004, 9, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Daré, B.; Lagente, V.; Gicquel, T. Ethanol and its metabolites: Update on toxicity, benefits, and focus on immunomodulatory effects. Drug Metab. Rev. 2019, 51, 545–561. [Google Scholar] [CrossRef]
- Zhao, P.; Slattery, J.T. Effects of ethanol dose and ethanol withdrawal on rat liver mitochondrial glutathione: Implication of potentiated acetaminophen toxicity in alcoholics. Drug Metab. Dispos. 2002, 30, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Michna, E.; Duh, M.S.; Korves, C.; Dahl, J.L. Removal of opioid/acetaminophen combination prescription pain medications: Assessing the evidence for hepatotoxicity and consequences of removal of these medications. Pain Med. 2010, 11, 369–378. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhelail, M.A.; Hoppe, J.A.; Rhyee, S.H.; Heard, K.J. Clinical course of repeated supratherapeutic ingestion of acetaminophen. Clin. Toxicol. 2011, 49, 108–112. [Google Scholar] [CrossRef]
- Hodgman, M.J.; Garrard, A.R. A review of acetaminophen poisoning. Crit. Care Clin. 2012, 28, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Kuffner, E.K.; Dart, R.C.; Bogdan, G.M.; Hill, R.E.; Casper, E.; Darton, L. Effect of maximal daily doses of acetaminophen on the liver of alcoholic patients: A randomized, double-blind, placebo-controlled trial. Arch. Intern. Med. 2001, 161, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Rumack, B.H. Acetaminophen misconceptions. Hepatology 2004, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.E.; Dalhoff, K.; Poulsen, H.E. Acute versus chronic alcohol consumption in acetaminophen-induced hepatotoxicity. Hepatology 2002, 35, 876–882. [Google Scholar] [CrossRef]
- Prescott, L.F. Paracetamol, alcohol and the liver. Br. J. Clin. Pharmacol. 2000, 49, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Waring, W.S.; Stephen, A.F.; Malkowska, A.M.; Robinson, O.D. Acute ethanol coingestion confers a lower risk of hepatotoxicity after deliberate acetaminophen overdose. Acad. Emerg. Med. 2008, 15, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.M.; Palmer, J.L.; Ippolito, J.A.; Curtis, B.J.; Choudhry, M.A.; Kovacs, E.J. Intoxication by intraperitoneal injection or oral gavage equally potentiates postburn organ damage and inflammation. Mediat. Inflamm. 2013, 2013, 971481. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Wang, W.; Eberhardt, A.; Vinitsky, H.S.; Reeves, B.C.; Peng, S.; Lou, N.; Hussain, R.; Nedergaard, M. Beneficial effects of low alcohol exposure, but adverse effects of high alcohol intake on glymphatic function. Sci. Rep. 2018, 8, 2246. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.X.; Kaplowitz, N. Role of innate immunity in acetaminophen-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 2006, 2, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [CrossRef]
- Yohe, H.C.; O’Hara, K.A.; Hunt, J.A.; Kitzmiller, T.J.; Wood, S.G.; Bement, J.L.; Bement, W.J.; Szakacs, J.G.; Wrighton, S.A.; Jacobs, J.M.; et al. Involvement of toll-like receptor 4 in acetaminophen hepatotoxicity. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1269–G1279. [Google Scholar] [CrossRef] [Green Version]
- Martin-Murphy, B.V.; Holt, M.P.; Ju, C. The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol. Lett. 2010, 192, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Williams, C.D.; Ramachandran, A.; Bajt, M.L. Acetaminophen hepatotoxicity and repair: The role of sterile inflammation and innate immunity. Liver Int. 2012, 32, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.E.; McKenzie, T.J.; Lillegard, J.B.; Yu, Y.; Juskewitch, J.E.; Nedredal, G.I.; Brunn, G.J.; Yi, E.S.; Malhi, H.; Smyrk, T.C.; et al. Role of Kupffer cells and toll-like receptor 4 in acetaminophen-induced acute liver failure. J. Surg. Res. 2013, 180, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Mossanen, J.C.; Tacke, F. Acetaminophen-induced acute liver injury in mice. Lab. Anim. 2015, 49 (Suppl. 1), 30–36. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Zheng, Z.; Shi, L.; Jin, Y.; Ji, L. Natural Polyphenol Chlorogenic Acid Protects Against Acetaminophen-Induced Hepatotoxicity by Activating ERK/Nrf2 Antioxidative Pathway. Toxicol. Sci. 2018, 162, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New Insights into Protein Kinase B/Akt Signaling: Role of Localized Akt Activation and Compartment-Specific Target Proteins for the Cellular Radiation Response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, S.; Cheng, H.; Lv, H.; Cheng, G.; Ci, X. Nrf2-mediated liver protection by esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3beta pathway. Free Radic. Biol. Med. 2016, 101, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Aliyu, N.O.; Ajala-Lawal, R.A.; Ajiboye, T.O. Lophirones B and C halt acetaminophen hepatotoxicity by upregulating redox transcription factor Nrf-2 through Akt, PI3K, and PKC pathways. J. Biochem. Mol. Toxicol. 2018, 32, e22055. [Google Scholar] [CrossRef] [PubMed]
- Schiødt, F.V.; Lee, W.M.; Bondesen, S.; Ott, P.; Christensen, E. Influence of acute and chronic alcohol intake on the clinical course and outcome in acetaminophen overdose. Aliment. Pharmacol. Ther. 2002, 16, 707–715. [Google Scholar] [CrossRef]
- Liao, C.C.; Day, Y.J.; Lee, H.C.; Liou, J.T.; Chou, A.H.; Liu, F.C. Baicalin attenuates IL-17-mediated acetaminophen-induced liver injury in a mouse model. PLoS ONE 2016, 11, e0166856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pösö, H.; Väänänen, H.; Salaspuro, M.P.; Pösö, A.R. Effects of ethanol on liver regeneration after partial hepatectomy in rats. Med. Biol. 1980, 58, 329–336. [Google Scholar] [PubMed]
- Diehl, A.M.; Thorgeirsson, S.S.; Steer, C.J. Ethanol inhibits liver regeneration in rats without reducing transcripts of key protooncogenes. Gastroenterology 1990, 99, 1105–1112. [Google Scholar] [CrossRef]
- Tuoi Do, T.H.; Gaboriau, F.; Ropert, M.; Moirand, R.; Cannie, I.; Brissot, P.; Loréal, O.; Lescoat, G. Ethanol effect on cell proliferation in the human hepatoma HepaRG cell line: Relationship with iron metabolism. Alcohol. Clin. Exp. Res. 2011, 35, 408–419. [Google Scholar] [CrossRef]
- Zhang, M.; Gong, Y.; Corbin, I.; Mellon, A.; Choy, P.; Uhanova, J.; Minuk, G.Y. Light ethanol consumption enhances liver regeneration after partial hepatectomy in rats. Gastroenterology 2000, 119, 1333–1339. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.-C.; Yu, H.-P.; Liao, C.-C.; Chou, A.-H.; Lee, H.-C. The Timing and Effects of Low-Dose Ethanol Treatment on Acetaminophen-Induced Liver Injury. Life 2021, 11, 1094. https://doi.org/10.3390/life11101094
Liu F-C, Yu H-P, Liao C-C, Chou A-H, Lee H-C. The Timing and Effects of Low-Dose Ethanol Treatment on Acetaminophen-Induced Liver Injury. Life. 2021; 11(10):1094. https://doi.org/10.3390/life11101094
Chicago/Turabian StyleLiu, Fu-Chao, Huang-Ping Yu, Chia-Chih Liao, An-Hsun Chou, and Hung-Chen Lee. 2021. "The Timing and Effects of Low-Dose Ethanol Treatment on Acetaminophen-Induced Liver Injury" Life 11, no. 10: 1094. https://doi.org/10.3390/life11101094
APA StyleLiu, F. -C., Yu, H. -P., Liao, C. -C., Chou, A. -H., & Lee, H. -C. (2021). The Timing and Effects of Low-Dose Ethanol Treatment on Acetaminophen-Induced Liver Injury. Life, 11(10), 1094. https://doi.org/10.3390/life11101094