
Molecular Pathogenesis and the Possible Role of Mitochondrial Heteroplasmy in Thoracic Aortic Aneurysm

 ,
,  ,
,  , , and
, , and 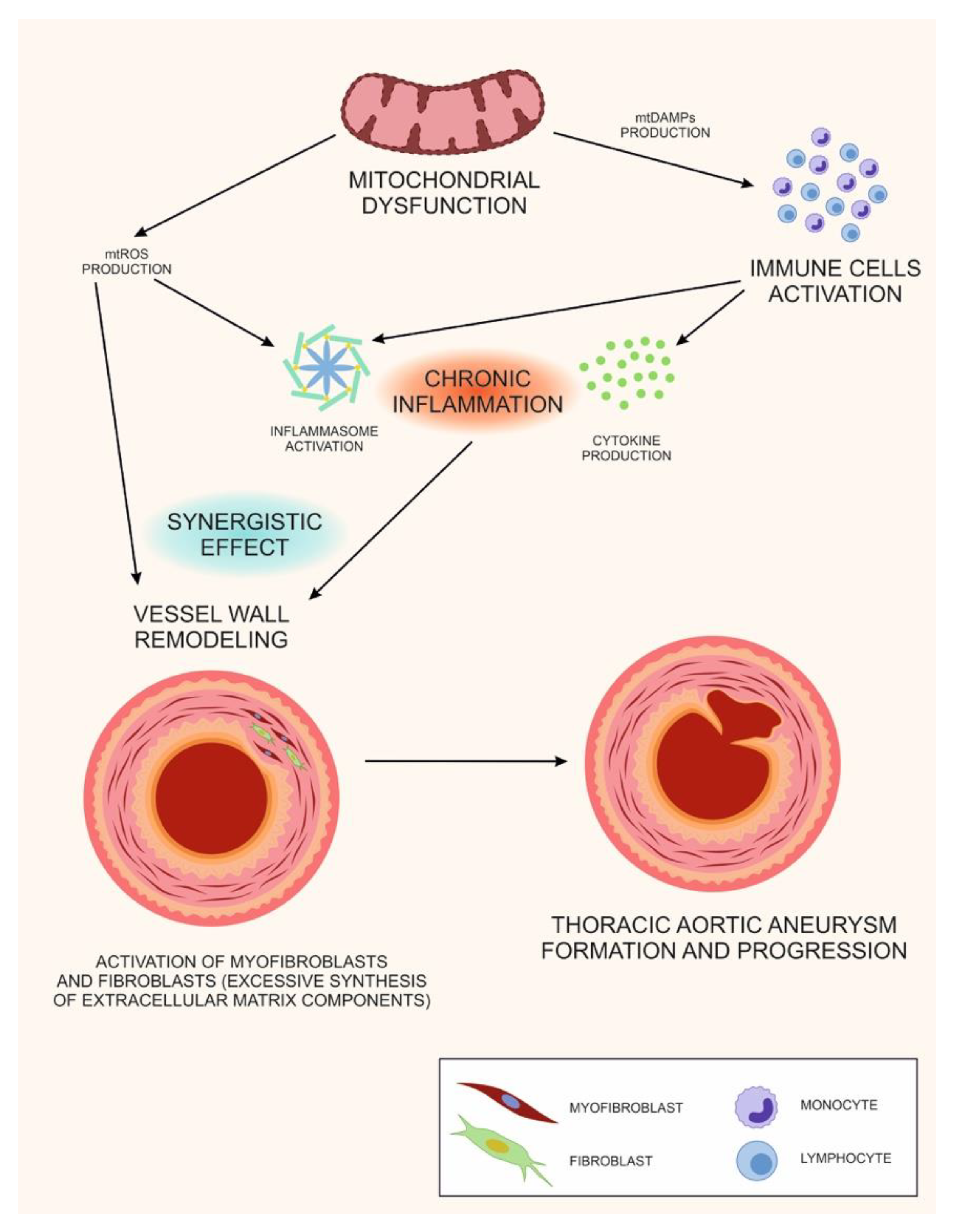{kind=link}
Abstract
:1. Introduction
2. Aortic Aneurysm Etiology and Pathogenesis
3. The Role of Mitochondrial DNA Heteroplasmy in Pathology Development
4. The Mitochondrion at the Crossroads of Metabolism, Inflammation and Immune Response
5. Molecular Mechanisms of Fibrogenesis and Mitochondrial Dysfunction
6. The Double Role of Mitochondrial Dysfunction in TAA: A Unifying Hypothesis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Safi, H.J.; Miller, C.C., 3rd; Estrera, A.L.; Huynh, T.T.; Rubenstein, F.S.; Subramaniam, M.H.; Buja, L.M. Staged Repair of Extensive Aortic Aneurysms. Circulation 2001, 104, 2938–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homme, J.L.; Aubry, M.-C.; Edwards, W.D.; Bagniewski, S.M.; Shane Pankratz, V.; Kral, C.A.; Tazelaar, H.D. Surgical pathology of the ascending aorta: A clinicopathologic study of 513 cases. Am. J. Surg. Pathol. 2006, 30, 1159–1168. [Google Scholar] [CrossRef]
- Lederle, F.A. In the clinic. Abdominal aortic aneurysm. Ann. Intern. Med. 2009, 150, ITC5-1. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Nair, C.K. Clinical, diagnostic, and management perspectives of aortic dissection. Chest 2002, 122, 311–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaideeswar, P.; Dixit, V.; Butany, J.; David, T.E.; Feindel, C. Surgical pathology of chronic ascending aortic dissections. Pathol. 2008, 40, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.V.; Isotalo, P.A.; Weyand, C.M.; Edwards, W.D.; Aubrym, M.-C.; Tazelaar, H.D. Surgical pathology of noninfectious ascending aortitis: A study of 45 cases with emphasis on an isolated variant. Am. J. Surg. Pathol. 2006, 30, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Schlatmann, T.J.; Becker, A.E. Pathogenesis of dissecting aneurysm of aorta: Comparative histopathologic study of significance of medial changes. Am. J. Cardiol. 1977, 39, 21–26. [Google Scholar] [CrossRef]
- Meyer, W.W.; Walsh, S.Z.; Lind, J. Functional Morphology of Human Arteries during Fetal and Postnatal Develop-Ment. In Structure and Function of the Circulation; Springer: Boston, MA, USA, 1980; pp. 95–379. [Google Scholar]
- Fritze, O.; Romero, B.; Schleicher, M.; Jacob, M.P.; Oh, D.-Y.; Starcher, B.; Schenke-Layland, K.; Bujan, J.; Stock, U.A. Age-Related Changes in the Elastic Tissue of the Human Aorta. J. Vasc. Res. 2012, 49, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.E.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar]
- Bickerstaff, L.K.; Pairolero, P.C.; Hollier, L.H.; Melton, L.J.; Van Peenen, H.J.; Cherry, K.J.; Joyce, J.W.; Lie, J.T. Thoracic aortic aneurysms: A population-Based study. Surgery 1982, 92, 1103–1108. [Google Scholar] [PubMed]
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Chute, E.P.; Littooy, F.N.; Bandyk, D.; Krupski, W.C.; Barone, G.W.; Acher, C.W.; Ballard, D.J. Prevalence and associations of ab-dominal aortic aneurysm detected through screening. Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Group. Ann. Intern. Med. 1997, 126, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Clouse, W.D.; Hallett, J.W.; Schaff, H.V.; Gayari, M.M.; Ilstrup, D.M.; Melton, L.J. Improved Prognosis of Thoracic Aortic Aneurysms: A Population-Based Study. Surv. Anesthesiol. 1999, 43, 198–199. [Google Scholar] [CrossRef]
- Moritz, A.R. Medionecrosis Aortae Idiopathica Cystica*. Am. J. Pathol. 1932, 8, 717–734.3. [Google Scholar]
- Dare, A.J.; Veinot, J.P.; Edwards, W.D.; Tazelaar, H.D.; Schaff, H.V. New observations on the etiology of aortic valve disease: A surgical pathologic study of 236 cases from 1990. Hum. Pathol. 1993, 24, 1330–1338. [Google Scholar] [CrossRef]
- Mayer, E.D.; Ruffmann, K.; Saggau, W.; Butzmann, B.; Bernhardt-Mayer, K.; Schatton, N.; Schmitz, W. Ruptured aneurysms of the sinus of Valsalva. Ann. Thorac. Surg. 1986, 42, 81–85. [Google Scholar] [CrossRef]
- Hoey, E.T.D.; Kanagasingam, A.; Sivananthan, M.U. Sinus of Valsalva Aneurysms: Assessment with Cardiovascular MRI. Am. J. Roentgenol. 2010, 194, W495–W504. [Google Scholar] [CrossRef] [PubMed]
- Grabenwöger, M.; Hutschala, D.; Ehrlich, M.P.; Cartes-Zumelzu, F.; Thurnher, S.; Lammer, J.; Wolner, E.; Havel, M. Thoracic aortic aneurysms: Treatment with endovascular self-Expandable stent grafts. Ann. Thorac. Surg. 2000, 69, 441–445. [Google Scholar] [CrossRef]
- Cook, J.R.; Carta, L.; Galatioto, J.; Ramirez, F. Cardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypes. Clin. Genet. 2015, 87, 11–20. [Google Scholar] [CrossRef]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 Antagonist, Prevents Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neptune, E.R.; Frischmeyer, P.A.; Arking, D.E.; Myers, L.; Bunton, T.E.; Gayraud, B.; Ramirez, F.; Sakai, L.Y.; Dietz, H.C. Dysregulation of TGF-beta activation con-tributes to pathogenesis in Marfan syndrome. Nat. Genet. 2003, 33, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.M.; Cheng, A.; Myers, L.A.; Martinez-Murillo, F.; Jie, C.; Bedja, D.; Gabrielson, K.L.; Hausladen, J.M.; Mecham, R.P.; Judge, D.P.; et al. TGF-Beta-Dependent pathogenesis of mitral valve pro-Lapse in a mouse model of Marfan syndrome. J. Clin. Investig. 2004, 114, 1586–1592. [Google Scholar] [CrossRef] [Green Version]
- Jondeau, G.; Boileau, C. Familial thoracic aortic aneurysms. Curr Opin Cardiol. 2014, 29, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Flachskampf, F.A.; Daniel, W.G. Aortic dissection. Cardiol. Clin. 2000, 18, 807–817. [Google Scholar] [CrossRef]
- Hagan, P.G.; Nienaber, C.A.; Isselbacher, E.M.; Bruckman, D.; Karavite, D.J.; Russman, P.L.; Evangelista, A.; Fattori, R.; Suzuki, T.; Oh, J.K.; et al. The International Registry of Acute Aortic Dissection (IRAD): New insights into an old disease. JAMA 2000, 283, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Parakatselaki, M.-E.; Ladoukakis, E. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schröder, R.; Ni, S.; Madea, B.; Stoneking, M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. USA 2015, 112, 2491–2496. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Rothwell, R.; Vermaat, M.; Wachsmuth, M.; Schröder, R.; Laros, J.F.; van Oven, M.; de Bakker, P.I.; Bovenberg, J.A.; van Duijn, C.M.; et al. Transmission of human mtDNA heteroplasmy in the Genome of the Netherlands families: Support for a variable-size bottleneck. Genome Res. 2016, 26, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaarniranta, K.; Pawlowska, E.; Szczepańska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef] [Green Version]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef] [PubMed]
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front. Mol. Biosci. 2021, 8, 671908. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Machado, T.S.; Macabelli, C.H.; Collado, M.D.; Meirelles, F.V.; Guimarães, F.E.G.; Chiaratti, M.R. Evidence of Selection Against Damaged Mitochondria During Early Embryogenesis in the Mouse. Front. Genet. 2020, 11, 762. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.B.; Chinnery, P.F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2021, 22, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Dorn, G.W. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Y.; Wang, F.; Huang, T.; Fan, K.; Zhang, Y.; Zhong, J.; Cao, Q.; Chao, T.; Jia, J.; et al. Mitochondrial dynamics controls anti-Tumour innate immunity by regulating CHIP-IRF1 axis stability. Nat. Commun. 2017, 8, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; Van Der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- De SouzaBreda, C.N.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox. Biol. 2019, 26, 101255. [Google Scholar]
- McCommis, K.S.; Baines, C.P. The role of VDAC in cell death: Friend or foe? Biochim. Biophys. Acta 2012, 1818, 1444–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-KappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiq-uitous mam-Malian homologs of the transmembrane GTPase Fzo. J. Cell. Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef]
- Fritz, S.; Rapaport, D.; Klanner, E.; Neupert, W.; Westermann, B. Connection of the Mitochondrial Outer and Inner Membranes by Fzo1 Is Critical for Organellar Fusion. J. Cell Biol. 2001, 152, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galonek, H.L.; Hardwick, J.M. Upgrading the BCL-2 network. Nat. Cell Biol. 2006, 8, 1317–1319. [Google Scholar] [CrossRef]
- Youle, R.J. Cell biology. Cellular Demolition and the Rules of Engagement. Science 2007, 315, 776–777. [Google Scholar] [CrossRef] [PubMed]
- Mazunin, I.O.; Levitskii, S.A.; Patrushev, M.V.; Kamenski, P.A. Mitochondrial matrix processes. Biochemistry 2015, 80, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.G.; Kim, S.-M.; Kim, K.-P.; Lee, S.-H.; Moon, J.-Y. The Role of Inflammasome-Dependent and Inflam-masome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaamonde-García, C.; López-Armada, M.J. Role of mitochondrial dysfunction on rheumatic diseases. Biochem. Pharmacol. 2019, 165, 181–195. [Google Scholar] [CrossRef]
- Wu, D.; Ren, P.; Zheng, Y.; Zhang, L.; Xu, G.; Xie, W.; Lloyd, E.E.; Zhang, S.; Zhang, Q.; Curci, J.A.; et al. NLRP3 (Nucleotide Oligomerization Domain–Like Receptor Family, Pyrin Domain Containing 3)–Caspase-1 Inflammasome Degrades Contractile Proteins. Arter. Thromb. Vasc. Biol. 2017, 37, 694–706. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, A.L.; Elezaby, A.; Qin, F.; Behring, J.B.; Luptak, I.; Calamaras, T.D.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease. J. Am. Hear. Assoc. 2016, 5, e002555. [Google Scholar] [CrossRef] [Green Version]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.-Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The Inflammasomes: Guardians of the Body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Dela Cruz, C.S.; Kang, M.-J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic in-flammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Picca, A.; Lezza, A.M.S.; Leeuwenburgh, C.; Pesce, V.; Calvani, R.; Landi, F. Fueling Inflamm-Aging through Mi-tochondrial Dysfunction: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2017, 18, 933. [Google Scholar] [CrossRef]
- Bajwa, E.; Pointer, C.B.; Klegeris, A. The Role of Mitochondrial Damage-Associated Molecular Patterns in Chronic Neuroin-flammation. Mediators Inflamm. 2019, 2019, 4050796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G.; Dewald, O.; Xia, Y.; Ren, G.; Haudek, S.; Leucker, T.; Kraemer, D.; Taffet, G.; Rollins, B.J.; Entman, M.L. Critical Role of Monocyte Chemoattractant Protein-1/CC Chemokine Ligand 2 in the Pathogenesis of Ischemic Cardiomyopathy. Circulation 2007, 115, 584–592. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Nastase, M.V.; Zeng-Brouwers, J.; Wygrecka, M.; Schaefer, L. Targeting renal fibrosis: Mechanisms and drug delivery systems. Adv. Drug. Deliv. Rev. 2018, 129, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A New Classification System and Their Role in Immunity. Immun 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yun, J.S.; Han, D.; Yook, J.I.; Kim, H.S.; Cho, E.S. TGF-β Pathway in Salivary Gland Fibrosis. Int. J. Mol. Sci. 2020, 21, 9138. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; Sauer-Lehnen, S.; Treptau, J.; Janoschek, N.; Theuerkauf, I.; Buettner, R.; Gressner, A.M.; Weiskirchen, R. Adenoviral expression of a transforming growth factor-beta1 antisense mRNA is effective in preventing liver fibrosis in bile-duct ligated rats. BMC Gastroenterol. 2003, 3, 29. [Google Scholar] [CrossRef] [Green Version]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportu-nities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, Z.; Zheng, J.; Yao, P.; Yuan, Z.; Lv, X.; Zhao, J.; Wang, M. IL-33 Can Promote the Process of Pulmonary Fibrosis by Inducing the Imbalance Between MMP-9 and TIMP-Inflammation. Inflammation. 2018, 41, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Garbern, J.C.; Williams, J.; Kristl, A.C.; Malick, A.; Rachmin, I.; Gaeta, B.; Ahmed, N.; Vujic, A.; Libby, P.; Lee, R.T. Dysregulation of IL-33/ST2 signaling and myocardial periarteriolar fibrosis. J. Mol. Cell. Cardiol. 2019, 128, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFβ1 on ROS generators, mediators and functional consequences. Redox Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Richter, K.; Kietzmann, T. Reactive oxygen species and fibrosis: Further evidence of a significant liaison. Cell Tissue Res. 2016, 365, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Manea, S.-A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chen, X.; Su, Y.; Paueksakon, P.; Hu, W.; Zhang, M.-Z.; Harris, R.C.; Blackwell, T.S.; Zent, R.; Pozzi, A. p47phox contributes to albuminuria and kidney fibrosis in mice. Kidney Int. 2015, 87, 948–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy-Marshman, H.; Quensel, K.; Shi-Wen, X.; Barnfield, R.; Kelly, J.; Peidl, A.; Stratton, R.J.; Leask, A. Antioxidants and NOX1/NOX4 inhibition blocks TGFβ1-induced CCN2 and α-SMA expression in dermal and gingival fibroblasts. PLoS ONE 2017, 12, e0186740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portelli, S.S.; Hambly, B.D.; Jeremy, R.W.; Robertson, E.N. Oxidative stress in genetically triggered thoracic aortic aneurysm: Role in pathogenesis and therapeutic opportunities. Redox Rep. 2021, 26, 45–52. [Google Scholar] [CrossRef]
- Soto, M.E.; Manzano-Pech, L.G.; Guarner-Lans, V.; Díaz-Galindo, J.A.; Vásquez, X.; Castrejón-Tellez, V.; Gamboa, R.; Huesca, C.; Fuentevilla-Alvárez, G.; Pérez-Torres, I. Oxidant/Antioxidant Profile in the Thoracic Aneurysm of Patients with the Loeys-Dietz Syndrome. Oxid. Med. Cell. Longev. 2020, 2020, 5392454. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucala, R.; Spiegel, L.A.; Chesney, J.; Hogan, M.; Cerami, A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med. 1994, 1, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, T.E.; Cowper, S.; Wu, S.-P.; Bockenstedt, L.K.; Bucala, R. Circulating fibrocytes: Collagen-secreting cells of the peripheral blood. Int. J. Biochem. Cell Biol. 2004, 36, 598–606. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Arciniegas, E.; Sutton, A.; Allen, T.; Schor, A. Transforming growth factor beta 1 promotes the differentiation of endothelial cells into smooth muscle-like cells in vitro. J. Cell Sci. 1992, 103, 521–529. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to Mesenchymal Transition (EndoMT) in the Patho-Genesis of Human Fibrotic Diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, S.; Wang, X.; Wang, Y.; Song, J.; Sun, C.; Chen, G.; Yang, G.; Tao, Y.; Hu, Y.; et al. Endothelial Cell-Derived SO2 Controls Endothelial Cell Inflammation, Smooth Muscle Cell Proliferation, and Collagen Synthesis to Inhibit Hypoxic Pulmonary Vascular Remodelling. Oxidative Med. Cell. Longev. 2021, 2021, 5577634. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jolly, A.J.; Strand, K.A.; Dubner, A.M.; Mutryn, M.F.; Moulton, K.S.; Nemenoff, R.A.; Majesky, M.W.; Weiser-Evans, M.C. Smooth muscle–derived progenitor cell myofibroblast differentiation through KLF4 downregulation promotes arterial remodeling and fibrosis. JCI Insight 2020, 5, e139445. [Google Scholar] [CrossRef]
- Douillet, C.D.; Velarde, V.; Christopher, J.T.; Mayfield, R.K.; Trojanowska, M.E.; Jaffa, A.A. Mechanisms by which bradykinin promotes fibrosis in vascular smooth muscle cells: Role of TGF-Beta and MAPK. Am. J. Physiol. Heart. Circ. Physiol. 2000, 279, H2829-37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Heydet, D.; Veldre, T.; Ghildyal, R. Transcriptomic changes during TGF-β-mediated differentiation of airway fibroblasts to myofibroblasts. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 20377. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-M.; Wei, C.-Y.; Wang, Q.; Wang, L.; Lu, L.; Qi, F.-Z. M2-polarized macrophages mediate wound healing by regulating connective tissue growth factor via AKT, ERK1/2, and STAT3 signaling pathways. Mol. Biol. Rep. 2021, 48, 6443–6456. [Google Scholar] [CrossRef]
- Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blanco-Colio, L.M. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int. J. Mol. Sci. 2021, 22, 7284. [Google Scholar] [CrossRef]
- Shimizu, K.; Mitchell, R.N.; Libby, P. Inflammation and Cellular Immune Responses in Abdominal Aortic Aneurysms. Arter. Thromb. Vasc. Biol. 2006, 26, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Infantes, D.; Nus, M.; Navas-Madroñal, M.; Fité, J.; Pérez, B.; Barros-Membrilla, A.; Soto, B.; Martínez-González, J.; Camacho, M.; Rodriguez, C.; et al. Oxidative Stress and Inflammatory Markers in Abdominal Aortic Aneurysm. Antioxidants 2021, 10, 602. [Google Scholar] [CrossRef] [PubMed]
- Postnov, A.; Suslov, A.; Sobenin, I.; Chairkin, I.; Sukhorukov, V.; Ekta, M.B.; Khotina, V.; Afanasiev, M.; Chumachenko, P.; Orekhov, A. Thoracic Aortic Aneurysm: Blood Pressure and Inflammation as Key Factors in the Development of Aneurysm Dissection. Curr. Pharm. Des. 2021, 27, 3122–3127. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Tarnopolsky, M.A. Exosomes as Mediators of the Systemic Adaptations to Endurance Exercise. Cold Spring Harb. Perspect. Med. 2018, 8, a029827. [Google Scholar] [CrossRef] [Green Version]
- Su, S.-A.; Xie, Y.; Fu, Z.; Wang, Y.; Wang, J.-A.; Xiang, M. Emerging role of exosome-mediated intercellular communication in vascular remodeling. Oncotarget 2017, 8, 25700–25712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Zhou, M.-X.; Zhou, F.-K.; Luo, X.-M.; Zhong, S.-X.; Zhou, Y.-F.; Qin, Y.-S.; Li, P.-P.; Qin, C. Exosome-Derived MiRNAs as Biomarkers of the Development and Progression of Intracranial Aneurysms. J. Atheroscler. Thromb. 2020, 27, 545–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerman, A.W.; Blanding, W.M.; Stroud, R.E.; Nadeau, E.K.; Mukherjee, R.; Ruddy, J.M.; Zile, M.R.; Ikonomidis, J.S.; Jones, J.A. Elevated Wall Tension Leads to Reduced miR-133a in the Thoracic Aorta by Exosome Release. J. Am. Hear. Assoc. 2019, 8, e010332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasta, S.; Agnese, V.; Gallo, A.; Cosentino, F.; Di Giuseppe, M.; Gentile, G.; Raffa, G.M.; Maalouf, J.F.; Michelena, H.I.; Bellavia, D.; et al. Shear Stress and Aortic Strain Associations with Biomarkers of Ascending Thoracic Aortic Aneurysm. Ann. Thorac. Surg. 2020, 110, 1595–1604. [Google Scholar] [CrossRef]
- Fernandez-García, C.E.; Burillo, E.; Lindholt, J.S.; Martinez-Lopez, D.; Pilely, K.; Mazzeo, C.; Michel, J.B.; Egido, J.; Garred, P.; Blanco-Colio, L.M.; et al. Association of ficolin-3 with abdominal aortic aneurysm presence and progression. J. Thromb. Haemost. 2017, 15, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Milewicz, D.M.; Trybus, K.M.; Guo, D.-C.; Sweeney, H.L.; Regalado, E.; Kamm, K. Altered Smooth Muscle Cell Force Generation as a Driver of Thoracic Aortic Aneurysms and Dissections. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Kulek, A.R.; Anzell, A.; Wider, J.M.; Sanderson, T.H.; Przyklenk, K. Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells 2020, 9, 214. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.D. The Mitochondria in Diabetic Heart Failure: From Pathogenesis to Therapeutic Promise. Antioxid. Redox Signal. 2015, 22, 1515–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassab, S.; Albalawi, Z.; Daghistani, H.; Kitmitto, A. Mitochondrial Arrest on the Microtubule Highway—A Feature of Heart Failure and Diabetic Cardiomyopathy? Front. Cardiovasc. Med. 2021, 8, 689101. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, N.; Lenaers, G.; Reynier, P.; Weissig, V.; Edeas, M. Mitochondrial Dysfunction in Mitochondrial Medicine: Current Limitations, Pitfalls, and Tomorrow. Methods Mol. Biol. 2021, 2276, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, D.; Zhao, L.; Wang, L.; Li, Y.; Cho, K.; Tao, C.; Jiang, B. Targeted depletion of monocyte/macrophage suppresses aortic dissection with the spatial regulation of MMP-9 in the aorta. Life Sci. 2020, 254, 116927. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Guo, D.-C.; Estrera, A.L.; Safi, H.J.; Huynh, T.T.; Yin, Z.; Cao, S.-N.; Lin, J.; Kurian, T.; Buja, L.M.; et al. Characterization of the inflammatory and apoptotic cells in the aortas of patients with ascending thoracic aortic aneurysms and dissections. J. Thorac. Cardiovasc. Surg. 2006, 131, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creamer, T.J.; Bramel, E.E.; MacFarlane, E.G. Insights on the Pathogenesis of Aneurysm through the Study of Hered-itary Aortopathies. Genes 2021, 12, 183. [Google Scholar] [CrossRef]
- Bunton, T.E.; Biery, N.J.; Myers, L.; Gayraud, B.; Ramirez, F.; Dietz, H.C. Phenotypic Alteration of Vascular Smooth Muscle Cells Precedes Elastolysis in a Mouse Model of Marfan Syndrome. Circ. Res. 2001, 88, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, E.; Foote, K.; Bennett, M. Mitochondrial function in thoracic aortic aneurysms. Cardiovasc. Res. 2018, 114, 1696–1698. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Grisanti, L.A. The Dynamic Interplay between Cardiac Inflammation and Fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suslov, A.V.; Afanasyev, M.A.; Degtyarev, P.A.; Chumachenko, P.V.; Ekta, M.B.; Sukhorukov, V.N.; Khotina, V.A.; Yet, S.-F.; Sobenin, I.A.; Postnov, A.Y. Molecular Pathogenesis and the Possible Role of Mitochondrial Heteroplasmy in Thoracic Aortic Aneurysm. Life 2021, 11, 1395. https://doi.org/10.3390/life11121395
Suslov AV, Afanasyev MA, Degtyarev PA, Chumachenko PV, Ekta MB, Sukhorukov VN, Khotina VA, Yet S-F, Sobenin IA, Postnov AY. Molecular Pathogenesis and the Possible Role of Mitochondrial Heteroplasmy in Thoracic Aortic Aneurysm. Life. 2021; 11(12):1395. https://doi.org/10.3390/life11121395
Chicago/Turabian StyleSuslov, A. V., M. A. Afanasyev, P. A. Degtyarev, P. V. Chumachenko, M. Bagheri Ekta, V. N. Sukhorukov, V. A. Khotina, S.-F. Yet, I. A. Sobenin, and A. Yu Postnov. 2021. "Molecular Pathogenesis and the Possible Role of Mitochondrial Heteroplasmy in Thoracic Aortic Aneurysm" Life 11, no. 12: 1395. https://doi.org/10.3390/life11121395
APA StyleSuslov, A. V., Afanasyev, M. A., Degtyarev, P. A., Chumachenko, P. V., Ekta, M. B., Sukhorukov, V. N., Khotina, V. A., Yet, S. -F., Sobenin, I. A., & Postnov, A. Y. (2021). Molecular Pathogenesis and the Possible Role of Mitochondrial Heteroplasmy in Thoracic Aortic Aneurysm. Life, 11(12), 1395. https://doi.org/10.3390/life11121395