
Integrative Analysis Identified Key Schizophrenia Risk Factors from an Abnormal Behavior Mouse Gene Set


Abstract
:1. Introduction
2. Materials and Methods
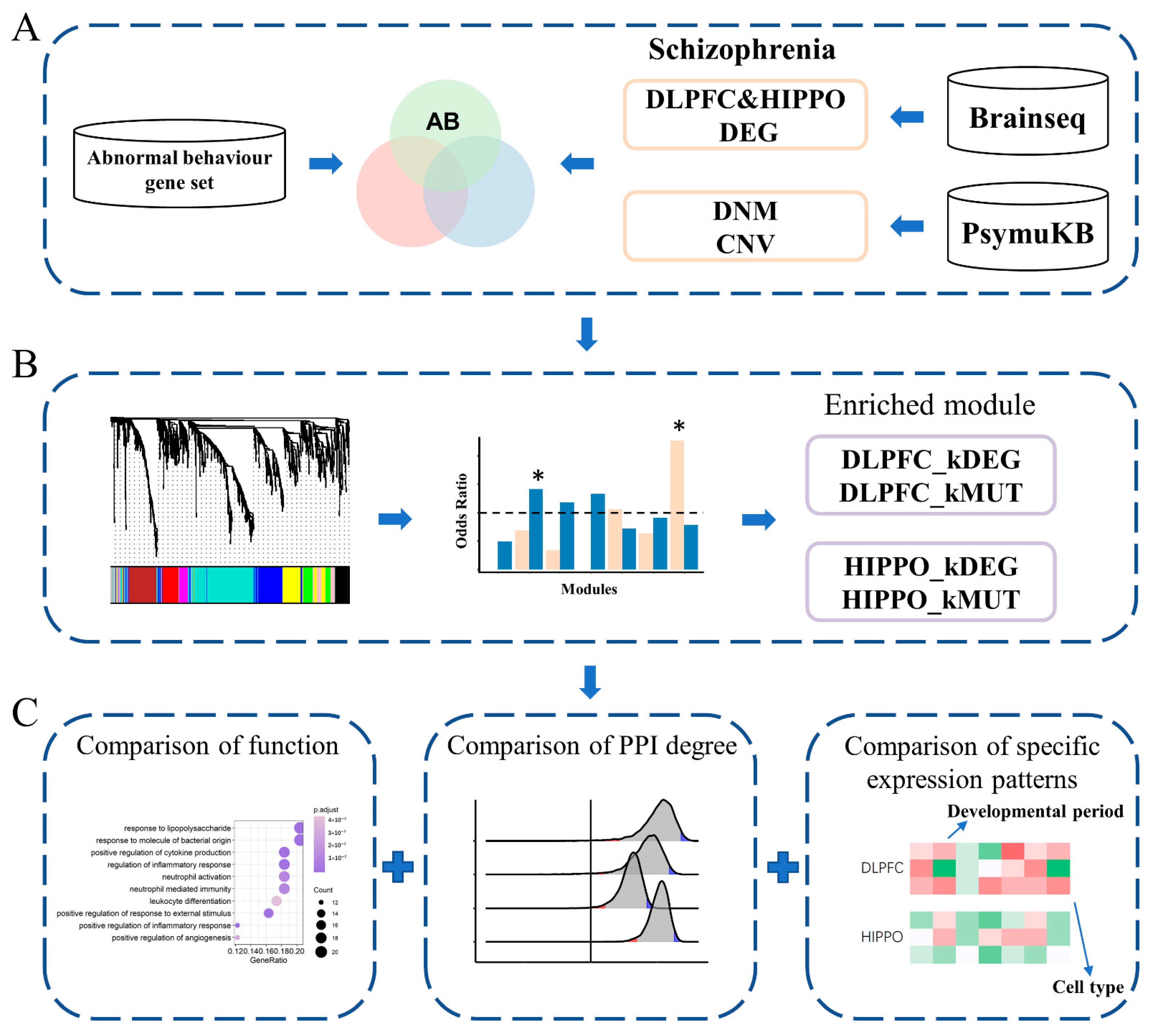
2.1. Data Collection and Filtration
2.2. Detection of Differentially Expressed Genes
2.3. Weighted Co-Expression Network Construction and Key Module Identification
2.4. Gene Ontology and Gene Set Enrichment Analyses
2.5. Analysis of Spatial/Cell-type Specific Expression of Genes
2.6. Brain-Expressed PPI Statistics for Disease Genes
3. Results
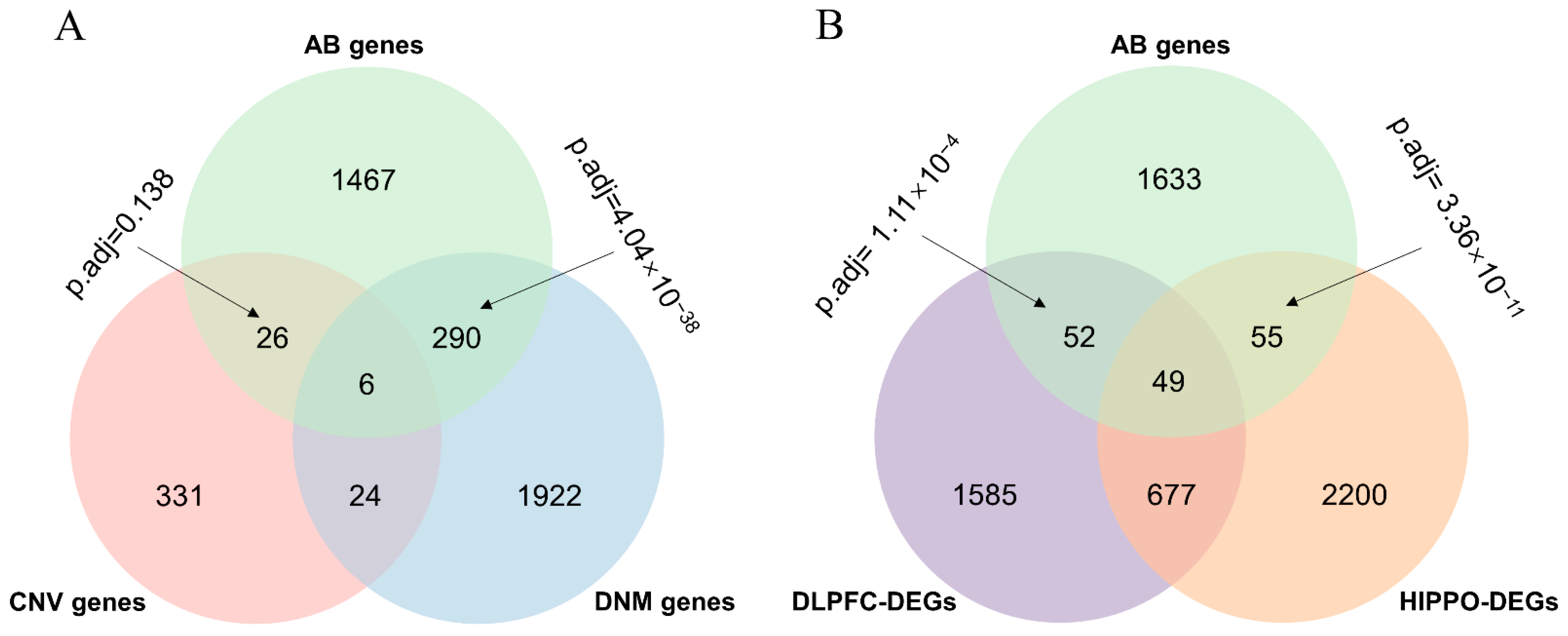
3.1. Abnormal Behavior Genes Were Significantly Enriched in SCZ Gene Sets
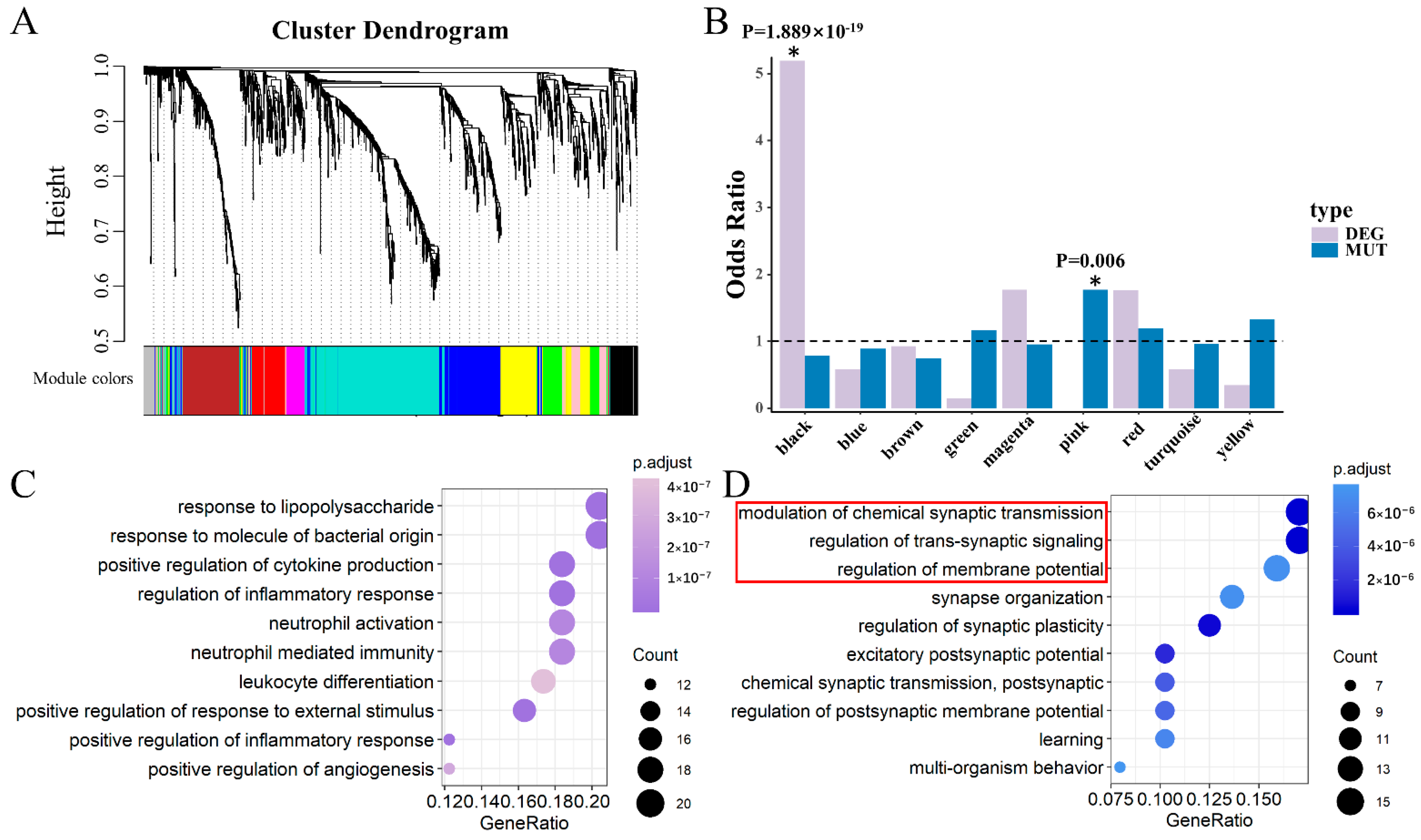
3.2. Two Significant Modules Were Identified in DLPFC
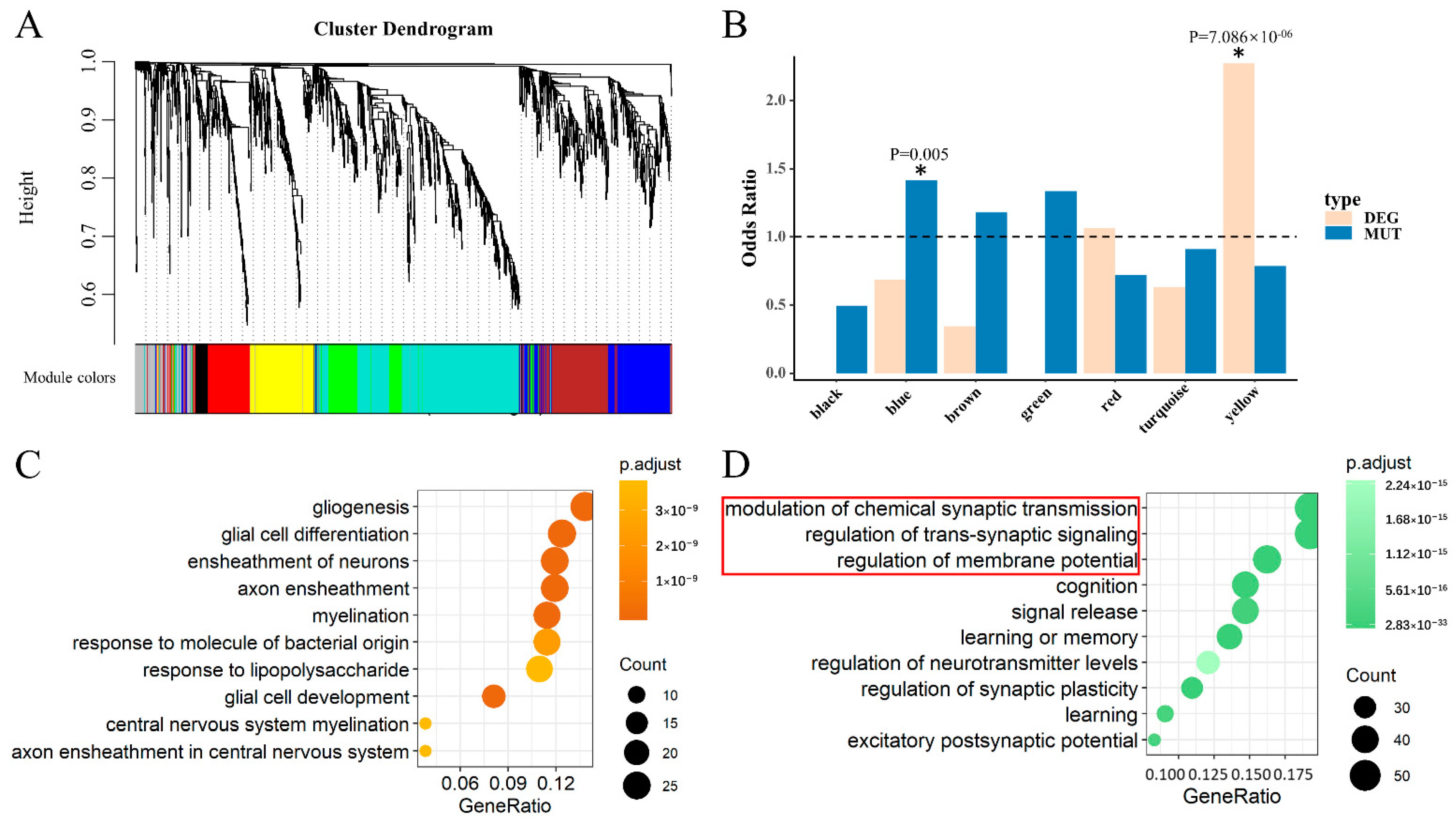
3.3. Two Significant Modules Were Identified in HIPPO
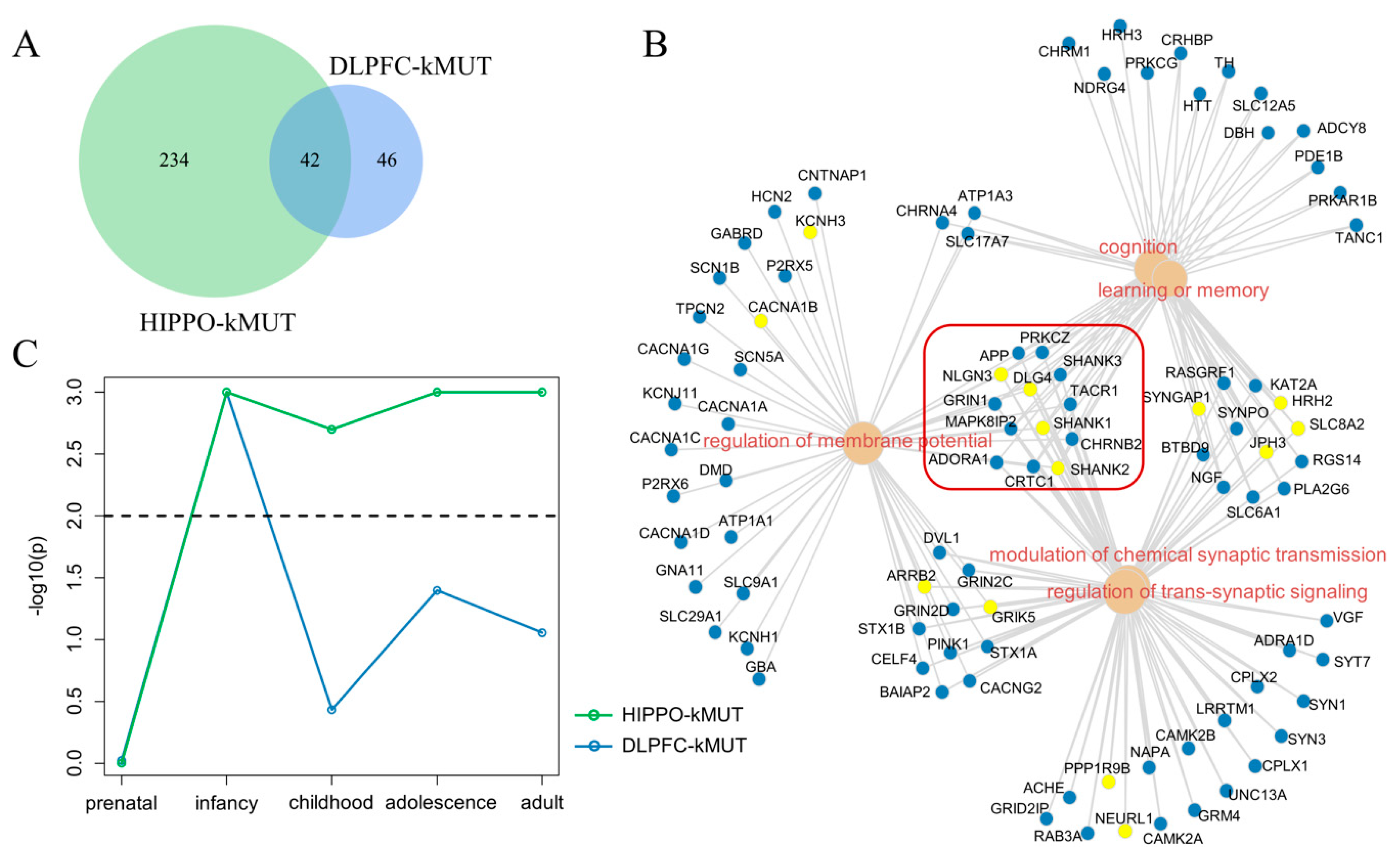
3.4. Comparison between DLPFC-kMUT and HIPPO-kMUT
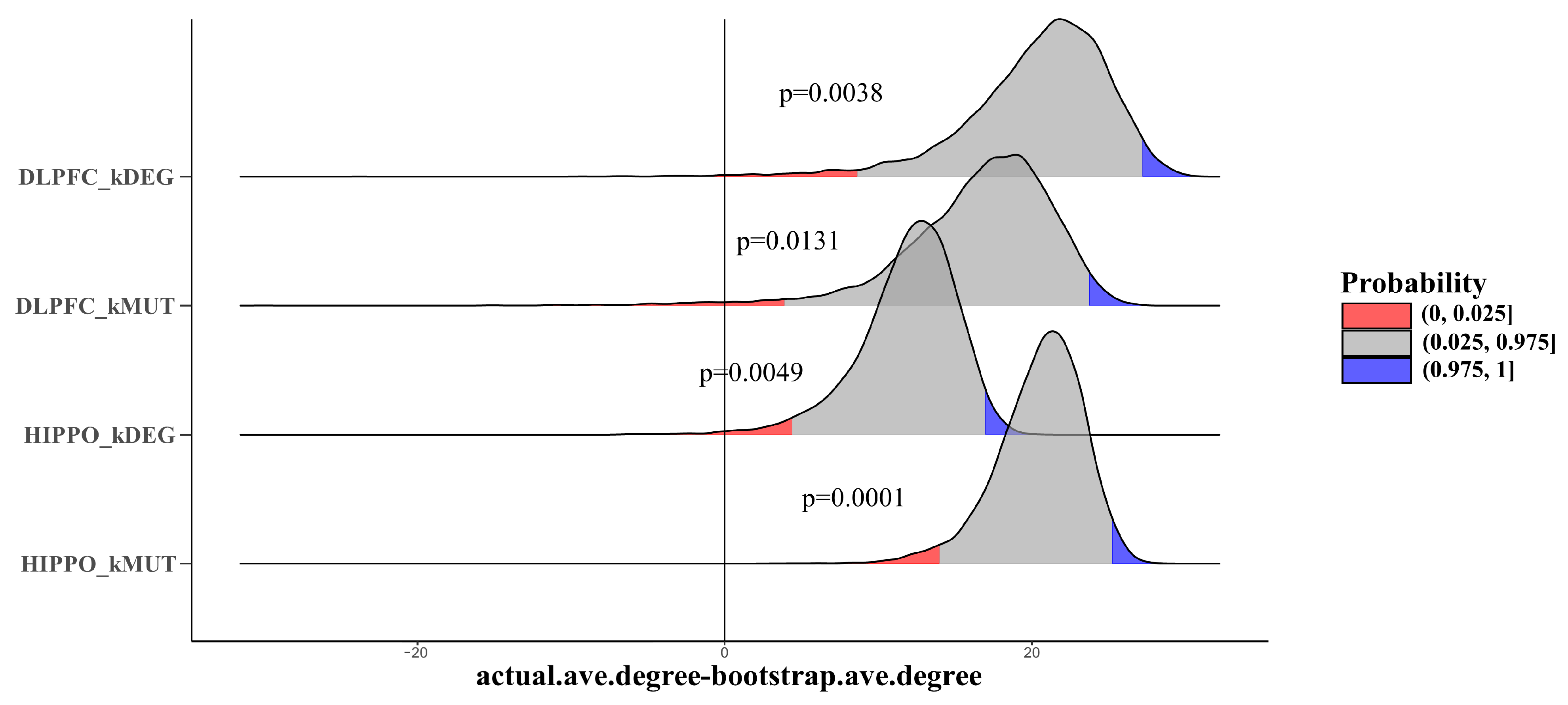
3.5. Larger Average PPI Degree Compared with Background
3.6. Different Cell-Type-Specific Expression Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Smeland, O.B.; Frei, O.; Dale, A.M.; Andreassen, O.A. The polygenic architecture of schizophrenia—rethinking pathogenesis and nosology. Nat. Rev. Neurol. 2020, 16, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Craddock, N.; O’Donovan, M.C.; Owen, M.J. The genetics of schizophrenia and bipolar disorder: Dissecting psychosis. J. Med. Genet. 2005, 42, 193. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.R.; Cherian, J.; Gohil, K.; Atkinson, D. Schizophrenia: Overview and treatment options. Pharm. Ther. 2014, 39, 638–645. [Google Scholar] [PubMed]
- Tandon, R.; Nasrallah, H.A.; Keshavan, M.S. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr. Res. 2009, 110, 1–23. [Google Scholar] [CrossRef]
- Blake, J.A.; Bult, C.J.; Eppig, J.T.; Kadin, J.A.; Richardson, J.E.; Mouse Genome Database Group. The Mouse Genome Database: Integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014, 42, D810–D817. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.; Chen, C.-Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avramopoulos, D. Recent Advances in the Genetics of Schizophrenia. Complex. Psychiatry 2018, 4, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Poterba, T.; Curtis, D.; Akil, H.; Al Eissa, M.; Barchas, J.D.; Bass, N.; Bigdeli, T.B.; Breen, G.; Bromet, E.J.; et al. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. Med. Rxiv. 2020. [Google Scholar] [CrossRef]
- Bassett, A.S.; Chow, E.W.C.; AbdelMalik, P.; Gheorghiu, M.; Husted, J.; Weksberg, R. The Schizophrenia Phenotype in 22q11 Deletion Syndrome. Am. J. Psychiatry 2003, 160, 1580–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howrigan, D.P.; Rose, S.A.; Samocha, K.E.; Fromer, M.; Cerrato, F.; Chen, W.J.; Churchhouse, C.; Chambert, K.; Chandler, S.D.; Daly, M.J.; et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 2020, 23, 185–193. [Google Scholar] [CrossRef]
- Girard, S.L.; Gauthier, J.; Noreau, A.; Xiong, L.; Zhou, S.; Jouan, L.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, E.; Diallo, O.; et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 2011, 43, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Yang, Z.; Hu, X.; Wu, H.-M.; Ni, P.; Ren, H.; Deng, W.; Li, M.; Ma, X.; et al. Increased co-expression of genes harboring the damaging de novo mutations in Chinese schizophrenic patients during prenatal development. Sci. Rep. 2015, 5, 18209. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Ionita-Laza, I.; Roos, J.L.; Boone, B.; Woodrick, S.; Sun, Y.; Levy, S.; Gogos, J.A.; Karayiorgou, M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012, 44, 1365–1369. [Google Scholar] [CrossRef] [Green Version]
- Guipponi, M.; Santoni, F.A.; Setola, V.; Gehrig, C.; Rotharmel, M.; Cuenca, M.; Guillin, O.; Dikeos, D.; Georgantopoulos, G.; Papadimitriou, G.; et al. Exome Sequencing in 53 Sporadic Cases of Schizophrenia Identifies 18 Putative Candidate Genes. PLoS ONE 2014, 9, e112745. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.N.; Guo, S.; Tan, X.; Wang, W.; Qian, W.; Song, W.; Wang, J.; Yu, S.; Wang, Z.; Cui, D.; et al. PsyMuKB: An Integrative De Novo Variant Knowledge Base for Developmental Disorders. Genom. Proteom. Bioinform. 2019, 17, 453–464. [Google Scholar] [CrossRef]
- Collado-Torres, L.; Burke, E.E.; Peterson, A.; Shin, J.; Straub, R.E.; Rajpurohit, A.; Semick, S.A.; Ulrich, W.S.; Price, A.J.; Valencia, C.; et al. Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron 2019, 103, 203–216. [Google Scholar] [CrossRef]
- Akbarian, S.; Liu, C.; Knowles, J.A.; Vaccarino, F.M.; Farnham, P.J.; Crawford, G.E.; Jaffe, A.E.; Pinto, D.; Dracheva, S.; Geschwind, D.H.; et al. The PsychENCODE project. Nat. Neurosci. 2015, 18, 1707–1712. [Google Scholar] [CrossRef] [Green Version]
- Schubert, C.R.; O’Donnell, P.; Quan, J.; Wendland, J.R.; Xi, H.S.; Winslow, A.R.; Domenici, E.; Essioux, L.; Kam-Thong, T.; Airey, D.C.; et al. BrainSeq: Neurogenomics to Drive Novel Target Discovery for Neuropsychiatric Disorders. Neuron 2015, 88, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Skene, N.G.; Grant, S.G.N. Identification of Vulnerable Cell Types in Major Brain Disorders Using Single Cell Transcriptomes and Expression Weighted Cell Type Enrichment. Front. Neurosci. 2016, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Ding, S.-L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030.e1016. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Reale, M.; Patruno, A.; De Lutiis, M.A.; Pesce, M.; Felaco, M.; Di Giannantonio, M.; Di Nicola, M.; Grilli, A. Dysregulation of chemo-cytokine production in schizophrenic patients versus healthy controls. BMC Neurosci. 2011, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Momtazmanesh, S.; Zare-Shahabadi, A.; Rezaei, N. Cytokine Alterations in Schizophrenia: An Updated Review. Front. Psychiatry 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lips, E.S.; Cornelisse, L.N.; Toonen, R.F.; Min, J.L.; Hultman, C.M.; Holmans, P.A.; O’Donovan, M.C.; Purcell, S.M.; Smit, A.B.; Verhage, M.; et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 2012, 17, 996–1006. [Google Scholar] [CrossRef]
- Lewis, D.A.; Lieberman, J.A. Catching Up on Schizophrenia: Natural History and Neurobiology. Neuron 2000, 28, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Tierney, A.L.; Nelson, C.A., 3rd. Brain Development and the Role of Experience in the Early Years. Zero Three 2009, 30, 9–13. [Google Scholar]
- Mirnics, K.; Middleton, F.A.; Marquez, A.; Lewis, D.A.; Levitt, P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 2000, 28, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Moon, A.L.; Haan, N.; Wilkinson, L.S.; Thomas, K.L.; Hall, J. CACNA1C: Association with Psychiatric Disorders, Behavior, and Neurogenesis. Schizophr. Bull. 2018, 44, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M.; et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, S.M.; Green, J.R.; Veeraragavan, S.; Yuva, L.; McCoy, A.; Wu, Y.; Warren, J.; Little, L.; Ji, D.; Cui, X.; et al. Fmr1 and Nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behav. Neurosci. 2014, 128, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Cheng, M.-C.; Qin, R.; Liao, D.-L.; Chen, T.-T.; Koong, F.-J.; Chen, G.; Chen, C.-H. Identification and functional characterization of rare mutations of the neuroligin-2 gene (NLGN2) associated with schizophrenia. Hum. Mol. Genet. 2011, 20, 3042–3051. [Google Scholar] [CrossRef]
- Yasuda, Y.; Hashimoto, R.; Yamamori, H.; Ohi, K.; Fukumoto, M.; Umeda-Yano, S.; Mohri, I.; Ito, A.; Taniike, M.; Takeda, M. Gene expression analysis in lymphoblasts derived from patients with autism spectrum disorder. Mol. Autism. 2011, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Meyer, U.; Feldon, J.; Dammann, O. Schizophrenia and autism: Both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr. Res. 2011, 69, 26R–33R. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Zhang, M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 2009, 10, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Feyder, M.; Karlsson, R.-M.; Mathur, P.; Lyman, M.; Bock, R.; Momenan, R.; Munasinghe, J.; Scattoni, M.L.; Ihne, J.; Camp, M.; et al. Association of Mouse Dlg4 (PSD-95) Gene Deletion and Human DLG4 Gene Variation with Phenotypes Relevant to Autism Spectrum Disorders and Williams’ Syndrome. Am. J. Psychiatry 2010, 167, 1508–1517. [Google Scholar] [CrossRef] [Green Version]
- Balan, S.; Yamada, K.; Hattori, E.; Iwayama, Y.; Toyota, T.; Ohnishi, T.; Maekawa, M.; Toyoshima, M.; Iwata, Y.; Suzuki, K.; et al. Population-specific haplotype association of the postsynaptic density gene DLG4 with schizophrenia, in family-based association studies. PLoS ONE 2013, 8, e70302. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.-C.; Lu, C.-L.; Luu, S.-U.; Tsai, H.-M.; Hsu, S.-H.; Chen, T.-T.; Chen, C.-H. Genetic and Functional Analysis of the DLG4 Gene Encoding the Post-Synaptic Density Protein 95 in Schizophrenia. PLoS ONE 2010, 5, e15107. [Google Scholar] [CrossRef]
- Kristiansen, L.V.; Beneyto, M.; Haroutunian, V.; Meador-Woodruff, J.H. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol. Psychiatry 2006, 11, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Kristiansen, L.V.; Meador-Woodruff, J.H. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr. Res. 2005, 78, 87–93. [Google Scholar] [CrossRef]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef] [Green Version]
- McAllister, A.K. Dynamic aspects of CNS synapse formation. Annu. Rev. Neurosci. 2007, 30, 425–450. [Google Scholar] [CrossRef] [Green Version]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 2008, 28, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Schmeisser, M.J.; Ey, E.; Wegener, S.; Bockmann, J.; Stempel, A.V.; Kuebler, A.; Janssen, A.L.; Udvardi, P.T.; Shiban, E.; Spilker, C.; et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 2012, 486, 256–260. [Google Scholar] [CrossRef]
- Won, H.; Lee, H.R.; Gee, H.Y.; Mah, W.; Kim, J.I.; Lee, J.; Ha, S.; Chung, C.; Jung, E.S.; Cho, Y.S.; et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 2012, 486, 261–265. [Google Scholar] [CrossRef]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism. 2010, 1, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [Green Version]
- Peykov, S.; Berkel, S.; Schoen, M.; Weiss, K.; Degenhardt, F.; Strohmaier, J.; Weiss, B.; Proepper, C.; Schratt, G.; Nöthen, M.M.; et al. Identification and functional characterization of rare SHANK2 variants in schizophrenia. Mol. Psychiatry 2015, 20, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Lennertz, L.; Wagner, M.; Wölwer, W.; Schuhmacher, A.; Frommann, I.; Berning, J.; Schulze-Rauschenbach, S.; Landsberg, M.W.; Steinbrecher, A.; Alexander, M.; et al. A promoter variant of SHANK1 affects auditory working memory in schizophrenia patients and in subjects clinically at risk for psychosis. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Champagne, N.; Lafreniere, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Cote, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [Green Version]
- Rual, J.-F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Li, S.; Armstrong, C.M.; Bertin, N.; Ge, H.; Milstein, S.; Boxem, M.; Vidalain, P.O.; Han, J.D.; Chesneau, A.; Hao, T.; et al. A map of the interactome network of the metazoan C. elegans. Science 2004, 303, 540–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, A.D.; Hescott, B.J.; Blumer, A.C.; Slonim, D.K. Connectedness of PPI network neighborhoods identifies regulatory hub proteins. Bioinformatics 2011, 27, 1135–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jonge, J.C.; Vinkers, C.H.; Hulshoff Pol, H.E.; Marsman, A. GABAergic Mechanisms in Schizophrenia: Linking Postmortem and In Vivo Studies. Front. Psychiatry 2017, 8, 118. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Wang, W.; Song, W.; Qian, W.; Lin, G.N. Integrative Analysis Identified Key Schizophrenia Risk Factors from an Abnormal Behavior Mouse Gene Set. Life 2021, 11, 172. https://doi.org/10.3390/life11020172
Chen M, Wang W, Song W, Qian W, Lin GN. Integrative Analysis Identified Key Schizophrenia Risk Factors from an Abnormal Behavior Mouse Gene Set. Life. 2021; 11(2):172. https://doi.org/10.3390/life11020172
Chicago/Turabian StyleChen, Miao, Weidi Wang, Weicheng Song, Wei Qian, and Guan Ning Lin. 2021. "Integrative Analysis Identified Key Schizophrenia Risk Factors from an Abnormal Behavior Mouse Gene Set" Life 11, no. 2: 172. https://doi.org/10.3390/life11020172
APA StyleChen, M., Wang, W., Song, W., Qian, W., & Lin, G. N. (2021). Integrative Analysis Identified Key Schizophrenia Risk Factors from an Abnormal Behavior Mouse Gene Set. Life, 11(2), 172. https://doi.org/10.3390/life11020172