
Animal Coronaviruses Induced Apoptosis

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
1.1. Coronaviruses (CoVs)
1.2. Host Cell Defense upon Infection: The Combined Effect of Antiviral Transcription and Programmed Cell Death in Establishment of Prolonged Immunity
1.2.1. Mechanisms of Apoptosis after Viral Infection
1.2.2. Components of Apoptotic Process
1.3. Viral Modulation of Apoptosis
2. Animal Coronaviruses (CoVs) and Apoptosis
2.1. Alphacoronaviruses
2.1.1. Porcine Epidemic Diarrhea Virus (PEDV)
2.1.2. Transmissible Gastroenteritis Virus (TGEV)
2.1.3. Swine Acute Diarrhea Syndrome Coronavirus (SADS-CoV)
2.1.4. Feline Coronavirus (FECV)
2.1.5. Canine Coronavirus (CCoV)
2.2. Betacoronaviruses
2.2.1. Μurine Hepatitis Virus (MHV)
2.2.2. Porcine Hemagglutinating Encephalomyelitis Virus (PHEV)
2.2.3. Equine Coronavirus (ECoV)
2.3. Gammacoronavirus
Avian Infectious Bronchitis Virus (IBV)
2.4. Deltacoronaviruses
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Cunningham, C.H.; Stuart, H.O. Cultivation of the virus of infectious bronchitis of chickens in embryonated chicken eggs. Am. J. Veter Res. 1947, 8, 209–212. [Google Scholar]
- Siddell, S.; Wege, H.; Ter Meulen, V. The Biology of Coronaviruses. J. Gen. Virol. 1983, 64, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [Green Version]
- Beloukas, A.; Magiorkinis, E.; Magiorkinis, G.; Zavitsanou, A.; Karamitros, T.; Hatzakis, A.; Paraskevis, D. Assessment of phylogenetic sensitivity for reconstructing HIV-1 epidemiological relationships. Virus Res. 2012, 166, 54–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef]
- Hay, S.; Kannourakis, G. A time to kill: Viral manipulation of the cell death program. J. Gen. Virol. 2002, 83, 1547–1564. [Google Scholar] [CrossRef]
- Upton, J.W.; Chan, F.K.M. Staying Alive: Cell Death in Antiviral Immunity. Mol. Cell 2014, 54, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.-T.; Chen, S.S.L. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell. Mol. Immunol. 2016, 13, 11–35. [Google Scholar] [CrossRef] [Green Version]
- González-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of Interferon-γ During Innate and Adaptive Immune Responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [Green Version]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nat. Cell Biol. 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Barber, G.N. The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ. 2005, 12, 563–570. [Google Scholar] [CrossRef]
- Kibler, K.V.; Shors, T.; Perkins, K.B.; Zeman, C.C.; Banaszak, M.P.; Biesterfeldt, J.; Langland, J.O.; Jacobs, B.L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 1997, 71, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Silverman, R.H. Viral Encounters with 2′,5′-Oligoadenylate Synthetase and RNase L during the Interferon Antiviral Response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvansakul, M.; Hinds, M.G. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013, 4, e909. [Google Scholar] [CrossRef]
- Tsukahara, T.; Kannagi, M.; Ohashi, T.; Kato, H.; Arai, M.; Nunez, G.; Iwanaga, Y.; Yamamoto, N.; Ohtani, K.; Nakamura, M.; et al. Induction of Bcl-x(L) expression by human T-cell leukemia virus type 1 Tax through NF-kappaB in apoptosis-resistant T-cell transfectants with Tax. J. Virol 1999, 73, 7981–7987. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; D’Aloja, P.; Schürmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-Kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Roizman, B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10410–10415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Davis, T.; Anderson, D.; Solam, L.; Beckmann, M.; Jerzy, R.; Dower, S.K.; Cosman, D.; Goodwin, R. A receptor for tumor necrosis factor defines an unusual family of cellular and viral proteins. Science 1990, 248, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollefson, A.E.; Scaria, A.; Hermiston, T.W.; Ryerse, J.S.; Wold, L.J.; Wold, W.S. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 1996, 70, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Prikhod’Ko, E.A.; Prikhod’Ko, G.G.; Siegel, R.M.; Thompson, P.; Major, M.E.; Cohen, J.I. The NS3 protein of hepatitis C virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 2004, 329, 53–67. [Google Scholar] [CrossRef]
- Pensaert, M.B.; De Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.J. Origin, Evolution, and Genotyping of Emergent Porcine Epidemic Diarrhea Virus Strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef] [Green Version]
- Egberink, H.F.; Ederveen, J.; Callebaut, P.; Horzinek, M.C. Characterization of the structural proteins of porcine epizootic diarrhea virus, strain CV777. Am. J. Veter Res. 1988, 49, 1320–1324. [Google Scholar]
- Kim, Y.; Lee, C. Porcine epidemic diarrhea virus induces caspase-independent apoptosis through activation of mitochondrial apoptosis-inducing factor. Virology 2014, 460-461, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Zhang, H.; Ding, Z.; Luo, R.; An, K.; Liu, L.; Bi, J.; Chen, H.; Xiao, S.; Fang, L. Proteome analysis of porcine epidemic diarrhea virus (PEDV)-infected Vero cells. Proteomics 2015, 15, 1819–1828. [Google Scholar] [CrossRef]
- Oh, C.; Kim, Y.; Chang, K.O. Caspase-mediated cleavage of nucleocapsid protein of a protease-independent porcine epidemic diarrhea virus strain. Virus Res. 2020, 285, 198026. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Z.; Li, J.; Gao, Y.; Zhou, L.; Ge, X.; Han, J.; Guo, X.; Yang, H. Porcine epidemic diarrhea virus S1 protein is the critical inducer of apoptosis. Virol. J. 2018, 15, 170. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Xu, Y.; Zhang, Q.; Yang, F.; Yin, Z.; Wang, L.; Li, Q. Porcine epidemic diarrhea virus infections induce apoptosis in Vero cells via a reactive oxygen species (ROS)/p53, but not p38 MAPK and SAPK/JNK signalling pathways. Veter. Microbiol. 2019, 232, 1–12. [Google Scholar] [CrossRef]
- Si, F.; Hu, X.; Wang, C.; Chen, B.; Wang, R.; Dong, S.; Yu, R.; Li, Z. Porcine Epidemic Diarrhea Virus (PEDV) ORF3 Enhances Viral Proliferation by Inhibiting Apoptosis of Infected Cells. Viruses 2020, 12, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Wu, H.; Huang, J.; Xu, Y.; Yang, F.; Zhang, Q.; Xu, X. Porcine epidemic diarrhea virus through p53-dependent pathway causes cell cycle arrest in the G0/G1 phase. Virus Res. 2018, 253, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Zhao, T.Q.; Xu, D.P.; Zhang, X.; Ji, C.J.; Zhang, D.L. The influence of porcine epidemic diarrhea virus on pig small intestine mucosal epithelial cell function. Arch. Virol. 2018, 164, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, B.; Chen, L.; Ma, Z.; He, K.; Fan, H. Differential Protein Analysis of IPEC-J2 Cells Infected with Porcine Epidemic Diarrhea Virus Pandemic and Classical Strains Elucidates the Pathogenesis of Infection. J. Proteome Res. 2017, 16, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Yin, L.; Pan, X.; Zhao, R.; Zhang, D. Porcine epidemic diarrhea virus infection blocks cell cycle and induces apoptosis in pig intestinal epithelial cells. Microb. Pathog. 2020, 147, 104378. [Google Scholar] [CrossRef]
- Eleouet, J.F.; Chilmonczyk, S.; Besnardeau, L.; Laude, H. Transmissible Gastroenteritis Coronavirus Induces Programmed Cell Death in Infected Cells through a Caspase-Dependent Pathway. J. Virol. 1998, 72, 4918–4924. [Google Scholar] [CrossRef] [Green Version]
- Yount, B.; Curtis, K.M.; Baric, R.S. Strategy for Systematic Assembly of Large RNA and DNA Genomes: Transmissible Gastroenteritis Virus Model. J. Virol. 2000, 74, 10600–10611. [Google Scholar] [CrossRef] [Green Version]
- Weingartl, H.M.; Derbyshire, J.B. Binding of porcine transmissible gastroenteritis virus by enterocytes from newborn and weaned piglets. Veter. Microbiol. 1993, 35, 23–32. [Google Scholar] [CrossRef]
- Kim, B.; Kim, O.; Tai, J.H.; Chae, C. Transmissible Gastroenteritis Virus Induces Apoptosis in Swine Testicular Cell Lines but not in Intestinal Enterocytes. J. Comp. Pathol. 2000, 123, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ding, L.; Li, Z.; Dai, M.; Zhao, X.; Li, W.; Du, Q.; Xu, X.; Tong, D. Transmissible gastroenteritis virus infection induces cell apoptosis via activation of p53 signalling. J. Gen. Virol. 2013, 94, 1807–1817. [Google Scholar] [CrossRef]
- Ding, L.; Zhao, X.; Huang, Y.; Du, Q.; Dong, F.; Zhang, H.; Song, X.; Zhang, W.; Tong, D. Regulation of ROS in transmissible gastroenteritis virus-activated apoptotic signaling. Biochem. Biophys. Res. Commun. 2013, 442, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, J.; Li, W.; Fang, Z.; Li, N.; Wu, S.; Li, J.; Hong, M. p53- and ROS-mediated AIF pathway involved in TGEV-induced apoptosis. J. Veter. Med Sci. 2018, 80, 1775–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Xu, X.; Huang, Y.; Li, Z.; Zhang, K.; Chen, G.; Yu, G.; Wang, Z.; Li, W.; Tong, D. Transmissible gastroenteritis virus infection induces apoptosis through FasL- and mitochondria-mediated pathways. Veter. Microbiol. 2012, 158, 12–22. [Google Scholar] [CrossRef]
- Ding, L.; Huang, Y.; Du, Q.; Dong, F.; Zhao, X.; Zhang, W.; Xu, X.; Tong, D. TGEV nucleocapsid protein induces cell cycle arrest and apoptosis through activation of p53 signaling. Biochem. Biophys. Res. Commun. 2014, 445, 497–503. [Google Scholar] [CrossRef]
- Eleouet, J.F.; Slee, E.A.; Saurini, F.; Castagne, N.; Poncet, D.; Garrido, C.; Solary, E.; Martin, S.J. The viral nucleocapsid protein of transmissible gastroenteritis coronavirus (TGEV) is cleaved by caspase-6 and -7 during TGEV-induced apoptosis. J. Virol. 2000, 74, 3975–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Song, X.; Bai, X.; Fei, N.; Huang, Y.; Zhao, Z.; Du, Q.; Zhang, H.; Zhang, L.; Tong, D. miR-27b attenuates apoptosis induced by transmissible gastroenteritis virus (TGEV) infection via targeting runt-related transcription factor 1 (RUNX1). PeerJ 2016, 4, e1635. [Google Scholar] [CrossRef]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nat. Cell Biol. 2018, 556, 255–258. [Google Scholar] [CrossRef]
- Zhang, J.; Han, Y.; Shi, H.; Chen, J.; Zhang, X.; Wang, X.; Zhou, L.; Liu, J.; Zhang, J.; Ji, Z.; et al. Swine acute diarrhea syndrome coronavirus-induced apoptosis is caspase- and cyclophilin D- dependent. Emerg. Microbes Infect. 2020, 9, 439–456. [Google Scholar] [CrossRef]
- Vennema, H.; Poland, A.; Foley, J.; Pedersen, N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 1998, 243, 150–157. [Google Scholar] [CrossRef] [Green Version]
- Kipar, A.; Meli, M.L. Feline infectious peritonitis: Still an enigma? Vet. Pathol. 2014, 51, 505–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, N.C. An update on feline infectious peritonitis: Virology and immunopathogenesis. Veter. J. 2014, 201, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Hohdatsu, T.; Hashida, Y.; Kaneko, Y.; Tanabe, M.; Koyama, H. A “possible” involvement of TNF-alpha in apoptosis induction in peripheral blood lymphocytes of cats with feline infectious peritonitis. Veter. Microbiol. 2007, 119, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Harun, M.S.R.; Kuan, C.O.; Selvarajah, G.T.; Wei, T.S.; Arshad, S.S.; Hair Bejo, M.; Omar, A.R. Transcriptional profiling of feline infectious peritonitis virus infection in CRFK cells and in PBMCs from FIP diagnosed cats. Virol. J. 2013, 10, 329. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Eckstrand, C.; Liu, H.; Pedersen, N.C. Characterization of peritoneal cells from cats with experimentally-induced feline infectious peritonitis (FIP) using RNA-seq. Veter. Res. 2018, 49, 81. [Google Scholar] [CrossRef] [Green Version]
- Drechsler, Y.; Vasconcelos, E.J.R.; Griggs, L.M.; Diniz, P.P.P.V.; Collisson, E. Host Gene Expression of Macrophages in Response to Feline Coronavirus Infection. Cells 2020, 9, 1431. [Google Scholar] [CrossRef]
- Shuid, A.N.; Safi, N.; Haghani, A.; Mehrbod, P.; Haron, M.S.; Tan, S.W.; Omar, A.R. Apoptosis transcriptional mechanism of feline infectious peritonitis virus infected cells. Apoptosis Int. J. Program. Cell Death 2015, 20, 1457–1470. [Google Scholar] [CrossRef]
- DeCaro, N.; Buonavoglia, C. An update on canine coronaviruses: Viral evolution and pathobiology. Veter. Microbiol. 2008, 132, 221–234. [Google Scholar] [CrossRef]
- Pratelli, A.; Martella, V.; DeCaro, N.; Tinelli, A.; Camero, M.; Cirone, F.; Elia, G.; Cavalli, A.; Corrente, M.; Greco, G.; et al. Genetic diversity of a canine coronavirus detected in pups with diarrhoea in Italy. J. Virol. Methods 2003, 110, 9–17. [Google Scholar] [CrossRef]
- Ruggieri, A.; Di Trani, L.; Gatto, I.; Franco, M.; Vignolo, E.; Bedini, B.; Elia, G.; Buonavoglia, C. Canine coronavirus induces apoptosis in cultured cells. Veter. Microbiol. 2007, 121, 64–72. [Google Scholar] [CrossRef] [PubMed]
- De Martino, L.; Marfé, G.; Longo, M.; Fiorito, F.; Montagnaro, S.; Iovane, V.; DeCaro, N.; Pagnini, U. Bid cleavage, cytochrome c release and caspase activation in canine coronavirus-induced apoptosis. Veter. Microbiol. 2010, 141, 36–45. [Google Scholar] [CrossRef]
- Sauve, A.A.; Wolberger, C.; Schramm, V.L.; Boeke, J.D. The Biochemistry of Sirtuins. Annu. Rev. Biochem. 2006, 75, 435–465. [Google Scholar] [CrossRef]
- Motta, M.C.; Divecha, N.; Lemieux, M.; Kamel, C.; Chen, D.; Gu, W.; Bultsma, Y.; McBurney, M.; Guarente, L. Mammalian SIRT1 Represses Forkhead Transcription Factors. Cell 2004, 116, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Marfé, G.; Tafani, M.; Fiorito, F.; Pagnini, U.; Iovane, G.; De Martino, L. Involvement of FOXO Transcription Factors, TRAIL-FasL/Fas, and Sirtuin Proteins Family in Canine Coronavirus Type II-Induced Apoptosis. PLoS ONE 2011, 6, e27313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheever, F.S.; Daniels, J.B.; Pappenheimer, A.M.; Bailey, O.T. A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin: I. Isolation and biological properties of the virus. J. Exp. Med. 1949, 90, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.J.; Weiss, S.R. Pathogenesis of murine coronavirus in the central nervous system. J. Neuroimmune Pharmacol. 2010, 5, 336–354. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.R.; Navas-Martin, S. Coronavirus Pathogenesis and the Emerging Pathogen Severe Acute Respiratory Syndrome Coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Dealbuquerque, N.; Baig, E.; Xuezhong, M.; Shalev, I.; Phillips, M.J.; Habal, M.; Leibowitz, J.; McGilvray, I.; Butany, J.; Fish, E.; et al. Murine Hepatitis Virus Strain 1 as a Model for Severe Acute Respiratory Distress Syndrome (Sars). Adv. Exp. Med. Biol. 2006, 581, 373–378. [Google Scholar] [CrossRef]
- Lavi, E.; Gilden, D.H.; Wroblewska, Z.; Rorke, L.B.; Weiss, S.R. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 1984, 34, 597–603. [Google Scholar] [CrossRef]
- Phillips, J.J.; Chua, M.M.; Lavi, E.; Weiss, S.R. Pathogenesis of Chimeric MHV4/MHV-A59 Recombinant Viruses: The Murine Coronavirus Spike Protein Is a Major Determinant of Neurovirulence. J. Virol. 1999, 73, 7752–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus Pathogenesis. Int. Rev. Cytol. 2011, 81, 85–164. [Google Scholar] [CrossRef]
- De Albuquerque, N.; Baig, E.; Ma, X.; Zhang, J.; He, W.; Rowe, A.; Habal, M.; Liu, M.; Shalev, I.; Downey, G.P.; et al. MurineHepatitis Virus Strain 1 Produces a Clinically Relevant Model of Severe Acute Respiratory Syndrome in A/J Mice. J. Virol. 2006, 80, 10382–10394. [Google Scholar] [CrossRef] [Green Version]
- Dveksler, G.S.; Pensiero, M.N.; Cardellichio, C.B.; Williams, R.K.; Jiang, G.S.; Holmes, K.V.; Dieffenbach, C.W. Cloning of the mouse hepatitis virus (MHV) receptor: Expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 1991, 65, 6881–6891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, T.J.; Weiss, S.R. Murine coronavirus neuropathogenesis: Determinants of virulence. J. NeuroVirol. 2010, 16, 427–434. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Chen, C.J.; Yu, X.; Leibowitz, J.L.; Makino, S. Induction of Apoptosis in Murine Coronavirus-Infected Cultured Cells and Demonstration of E Protein as an Apoptosis Inducer. J. Virol. 1999, 73, 7853–7859. [Google Scholar] [CrossRef] [Green Version]
- Belyavsky, M.; Belyavskaya, E.; Levy, G.A.; Leibowitz, J.L. Coronavirus MHV-3-Induced Apoptosis in Macrophages. Virology 1998, 250, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz, J.L.; Belyavskaya, E. Caspase Inhibitors Block MHV-3 Induced Apoptosis and Enhance Viral Replication and Pathogenicity. Adv. Exp. Med. Biol. 2001, 494, 109–114. [Google Scholar] [CrossRef]
- Yang, C.; Chen, Y.; Guo, G.; Li, H.; Cao, D.; Xu, H.; Guo, S.; Fei, L.; Yan, W.; Ning, Q.; et al. Expression of B and T lymphocyte attenuator (BTLA) in macrophages contributes to the fulminant hepatitis caused by murine hepatitis virus strain-3. Gut 2013, 62, 1204–1213. [Google Scholar] [CrossRef]
- Barac-Latas, V.; Suchanek, G.; Breitschopf, H.; Stuehler, A.; Wege, H.; Lassmann, H. Patterns of oligodendrocyte pathology in coronavirus-induced subacute demyelinating encephalomyelitis in the lewis rat. Glia 1997, 19, 1–12. [Google Scholar] [CrossRef]
- Wu, G.F.; Perlman, S. Macrophage Infiltration, but Not Apoptosis, Is Correlated with Immune-Mediated Demyelination following Murine Infection with a Neurotropic Coronavirus. J. Virol. 1999, 73, 8771–8780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, T.; Fu, L.; Lavi, E. Differential induction of apoptosis in demyelinating and nondemyelinating infection by mouse hepatitis virus. J. NeuroVirol. 2002, 8, 392–399. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X. Murine coronavirus-induced oligodendrocyte apoptosis is mediated through the activation of the Fas signaling pathway. Virology 2007, 360, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Youn, H.Y.; Hasegawa, A.; Nakayama, H.; Goto, N. Apoptotic Changes in the Thymus of Mice Infected with Mouse Hepatitis Virus, MHV-2. J. Veter. Med Sci. 1994, 56, 879–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Clin. Chem. 2006, 66, 193–292. [Google Scholar] [CrossRef]
- Quiroga, M.A.; Cappuccio, J.; Piñeyro, P.; Basso, W.; More, G.; Kienast, M.; Schonfeld, S.; Cancer, J.L.; Arauz, S.; Pintos, M.E.; et al. Hemagglutinating Encephalomyelitis Coronavirus Infection in Pigs, Argentina. Emerg. Infect. Dis. 2008, 14, 484–486. [Google Scholar] [CrossRef]
- Hara, Y.; Hasebe, R.; Sunden, Y.; Ochiai, K.; Honda, E.; Sakoda, Y.; Umemura, T. Propagation of Swine Hemagglutinating Encephalomyelitis Virus and Pseudorabies Virus in Dorsal Root Ganglia Cells. J. Veter. Med. Sci. 2009, 71, 595–601. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Zhao, K.; Wang, G.; Dong, B.; Zhao, J.; Tang, B.; Lu, H.; Gao, W.; Chang, L.; Jin, Z.; et al. Porcine hemagglutinating encephalomyelitis virus induces apoptosis in a porcine kidney cell line via caspase-dependent pathways. Virus Res. 2013, 176, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhao, K.; Zhao, J.; Lv, X.; Wang, G.; Lu, H.; Tang, B.; Li, Z.; Chang, L.; Jin, Z.; et al. Gene-expression patterns in the cerebral cortex of mice infected with porcine haemagglutinating encephalomyelitis virus detected using microarray. J. Gen. Virol. 2014, 95, 2192–2203. [Google Scholar] [CrossRef]
- Zhang, J.; Guy, J.S.; Snijder, E.J.; Denniston, D.A.; Timoney, P.J.; Balasuriya, U.B. Genomic characterization of equine coronavirus. Virology 2007, 369, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusterla, N.; Vin, R.; Leutenegger, C.; Mittel, L.D.; Divers, T.J. Equine coronavirus: An emerging enteric virus of adult horses. Equine Veter. Educ. 2016, 28, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Matsui, Y.; Miura, Y.; Sentsui, H. Equine coronavirus induces apoptosis in cultured cells. Veter. Microbiol. 2008, 129, 390–395. [Google Scholar] [CrossRef]
- Kinde, H.; Daft, B.M.; Castro, A.E.; Bickford, A.A.; Gelb, J., Jr.; Reynolds, B. Viral Pathogenesis of a Nephrotropic Infectious Bronchitis Virus Isolated from Commercial Pullets. Avian Dis. 1991, 35, 415–421. [Google Scholar] [CrossRef]
- Ahmed, Z.; Naeem, K.; Hameed, A. Detection and Seroprevalence of Infectious Bronchitis Virus Strains in Commercial Poultry in Pakistan. Poult. Sci. 2007, 86, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Sun, C.; Yan, B.; Zhang, X.; Wang, Y.; Li, C.; Zhang, Q.; Ma, Y.; Shao, Y.; Liu, Q.; et al. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011, 11, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases 2016, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, H.Y.; Liu, D.X. Induction of Caspase-Dependent Apoptosis in Cultured Cells by the Avian Coronavirus Infectious Bronchitis Virus. J. Virol. 2001, 75, 6402–6409. [Google Scholar] [CrossRef] [Green Version]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007, 365, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Liao, Y.; Fang, S.; Tam, J.P.; Liu, D.X. Up-Regulation of Mcl-1 and Bak by Coronavirus Infection of Human, Avian and Animal Cells Modulates Apoptosis and Viral Replication. PLoS ONE 2012, 7, e30191. [Google Scholar] [CrossRef] [Green Version]
- Cong, F.; Liu, X.; Han, Z.; Shao, Y.; Kong, X.; Liu, S. Transcriptome analysis of chicken kidney tissues following coronavirus avian infectious bronchitis virus infection. BMC Genom. 2013, 14, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, X.; Zhang, Z.; Li, J.; Zou, W.; Zeng, F.; Wang, H. Comparative transcriptome analysis reveals induction of apoptosis in chicken kidney cells associated with the virulence of nephropathogenic infectious bronchitis virus. Microb. Pathog. 2017, 113, 451–459. [Google Scholar] [CrossRef]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [Green Version]
- Fung, T.S.; Liu, D.X. Activation of the c-Jun NH2-terminal kinase pathway by coronavirus infectious bronchitis virus promotes apoptosis independently of c-Jun. Cell Death Dis. 2017, 8, 3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.X.; Liang, J.Q.; Zhu, Q.C.; Dai, G.; Li, S.; Fung, T.S.; Liu, D.X. Gammacoronavirus Avian Infectious Bronchitis Virus and Alphacoronavirus Porcine Epidemic Diarrhea Virus Exploit a Cell-Survival Strategy via Upregulation of cFOS to Promote Viral Replication. J. Virol. 2020, e02107-20. [Google Scholar] [CrossRef]
- Liang, J.Q.; Fang, S.; Yuan, Q.; Huang, M.; Chen, R.A.; Fung, T.S.; Liu, D.X. N-Linked glycosylation of the membrane protein ectodomain regulates infectious bronchitis virus-induced ER stress response, apoptosis and pathogenesis. Virology 2019, 531, 48–56. [Google Scholar] [CrossRef]
- Han, X.; Tian, Y.; Guan, R.; Gao, W.; Yang, X.; Zhou, L.; Wang, H. Infectious Bronchitis Virus Infection Induces Apoptosis during Replication in Chicken Macrophage HD11 Cells. Viruses 2017, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, C. Complete Genome Characterization of Korean Porcine Deltacoronavirus Strain KOR/KNU14-04/2014. Genome Announc. 2014, 2, e01191-14. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.; Hu, H.; Eyerly, B.; Lu, Z.; Chepngeno, J.; Saif, L.J. Pathogenicity of 2 Porcine Deltacoronavirus Strains in Gnotobiotic Pigs. Emerg. Infect. Dis. 2015, 21, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Hu, H.; Saif, L.J. Porcine deltacoronavirus induces apoptosis in swine testicular and LLC porcine kidney cell lines in vitro but not in infected intestinal enterocytes in vivo. Veter. Microbiol. 2016, 182, 57–63. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, C. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c -mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, L.; Zhou, P.; Zhang, Y.; Wei, Y.; Wang, Y.; Liu, X. Tandem Mass Tag-Based Quantitative Proteome Analysis of Porcine Deltacoronavirus (PDCoV)-Infected LLC Porcine Kidney Cells. ACS Omega 2020, 5, 21979–21987. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J. Initiator caspases in apoptosis signaling pathways. Apoptosis Int. J. Program. Cell Death 2002, 7, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Goldstaub, D.; Gradi, A.; Bercovitch, Z.; Grosmann, Z.; Nophar, Y.; Luria, S.; Sonenberg, N.; Kahana, C. Poliovirus 2A Protease Induces Apoptotic Cell Death. Mol. Cell. Biol. 2000, 20, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Barco, A.; Feduchi, E.; Carrasco, L. Poliovirus protease 3C(pro) kills cells by apoptosis. Virology 2000, 266, 352–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuconati, A.; Mukherjee, C.; Perez, D.; White, E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003, 17, 2922–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Chipuk, J.E.; Boyd, K.L.; Van De Velde, N.; Green, D.R.; McCullers, J.A. PB1-F2 Proteins from H5N1 and 20th Century Pandemic Influenza Viruses Cause Immunopathology. PLoS Pathog. 2010, 6, e1001014. [Google Scholar] [CrossRef] [Green Version]
- Jacotot, E.; Ferri, K.F.; El Hamel, C.; Brenner, C.; Druillennec, S.; Hoebeke, J.; Rustin, P.; Metivier, D.; Lenoir, C.; Geuskens, M.; et al. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J. Exp. Med. 2001, 193, 509–519. [Google Scholar] [CrossRef]
- Coffey, C.M.; Sheh, A.; Kim, I.S.; Chandran, K.; Nibert, M.L.; Parker, J.S.L. Reovirus Outer Capsid Protein μ1 Induces Apoptosis and Associates with Lipid Droplets, Endoplasmic Reticulum, and Mitochondria. J. Virol. 2006, 80, 8422–8438. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.X.; Tan, T.H.P.; Lee, M.J.R.; Tham, P.-Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of Apoptosis by the Severe Acute Respiratory Syndrome Coronavirus 7a Protein Is Dependent on Its Interaction with the Bcl-XL Protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Freundt, E.C.; Yu, L.; Goldsmith, C.S.; Welsh, S.; Cheng, A.; Yount, B.; Liu, W.; Frieman, M.B.; Buchholz, U.J.; Screaton, G.R.; et al. The Open Reading Frame 3a Protein of Severe Acute Respiratory Syndrome-Associated Coronavirus Promotes Membrane Rearrangement and Cell Death. J. Virol. 2010, 84, 1097–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, P.T.W.; Wong, C.H.; Au, T.C.C.; Chuck, C.P.; Kong, S.K.; Chan, P.K.S.; To, K.F.; Lo, A.W.I.; Chan, J.Y.W.; Suen, Y.K.; et al. The 3a protein of severe acute respiratory syndrome-associated coronavirus induces apoptosis in Vero E6 cells. J. Gen. Virol. 2005, 86, 1921–1930. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.-Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Zhang, Q.; Ke, H.; Blikslager, A.; Fujita, T.; Yoo, D. Type III Interferon Restriction by Porcine Epidemic Diarrhea Virus and the Role of Viral Protein nsp1 in IRF1 Signaling. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]



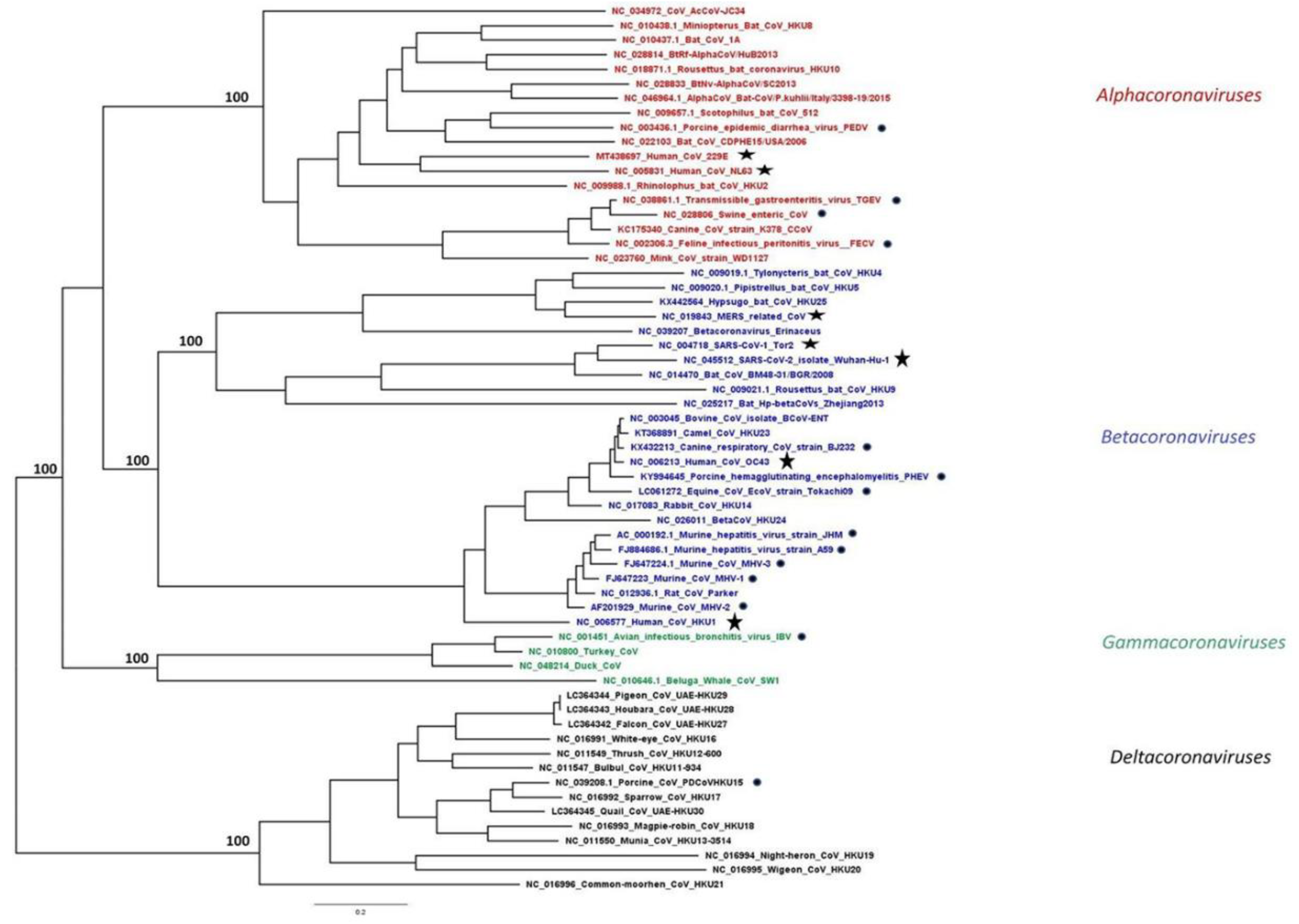{kind=link}
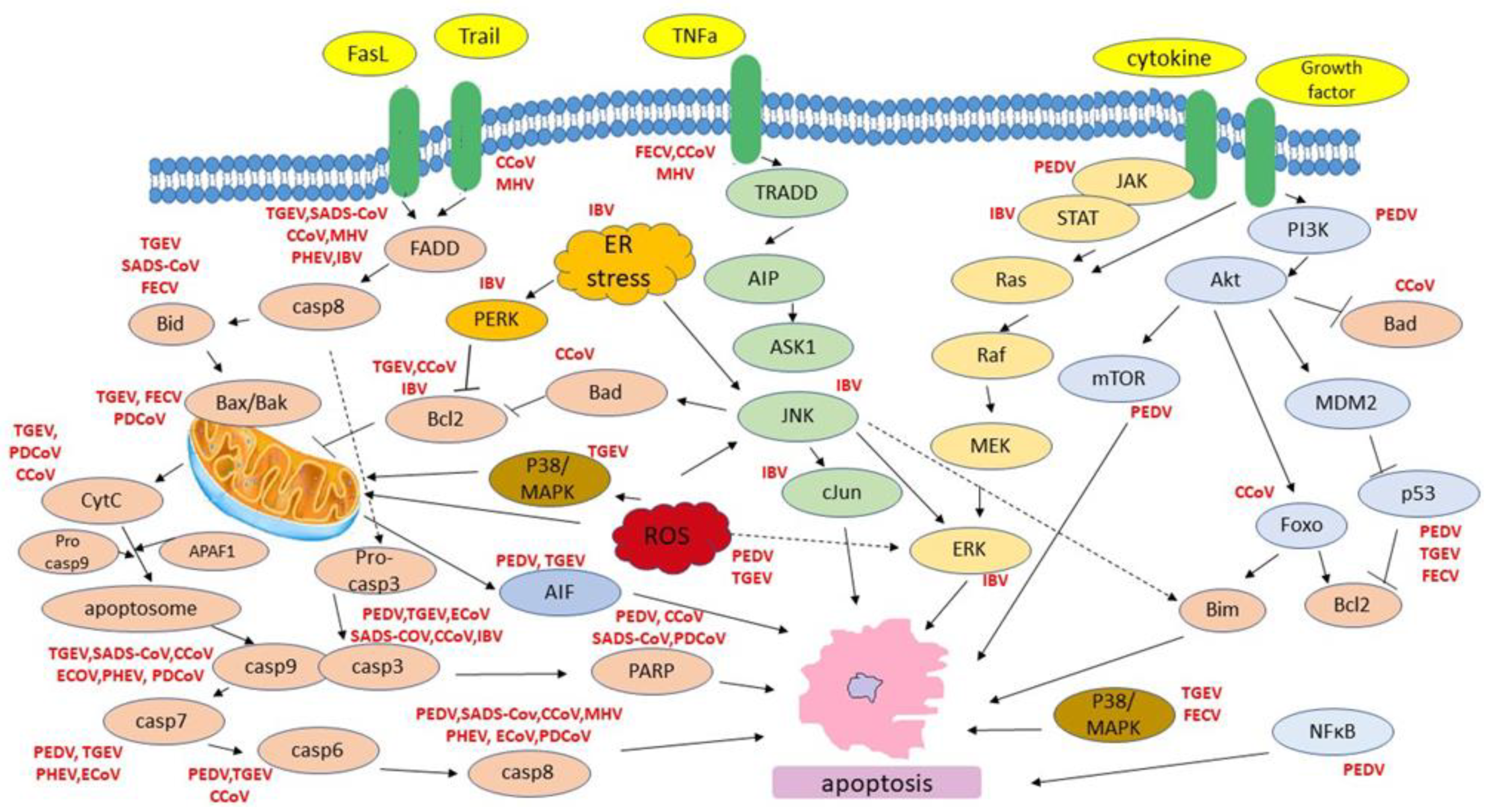{kind=link}
| Coronavirus | Strain | In Vitro/In Vivo | Apoptotic Pathway | Ref. |
|---|---|---|---|---|
| Alphacoronaviruses | ||||
| PEDV Host: swine | SM98-1 | Vero Tissue specimens from duodenum, jejunum and ileum | ↑AIF | [30] |
| AJ1102 | Vero | 12 apoptotic proteins | [31] | |
| 8aa and KD | Vero | caspase 6 or 7 | [32] | |
| CV777 or SM98 | Vero, Vero-E6, and Marc-145 cells | ↑caspase-3, ↑caspase-8, AIFM1 and PARP cleavage | [33] | |
| Shaanxi | Vero | ↑ p53, ↑ ROS | [34] | |
| DR13, CV777, rDR13att-ORF3CV777 rDR13att-ORF3NY rDR13att-∆ORF3 | LR7, Vero CCL-81 | ↓caspase-3 Cleavage | [35] | |
| CH/SXYL/2016 | Vero | G0/G1 arrest, ↑p53 | [36] | |
| CV777 | Vero, immortalized pig small intestinal mucosal epithelial cells | ↑apoptosis | [37] | |
| YC2014, CV777 | IPEC-J2 | ↑JAK-STAT, ↑NF-kB ↓PI3K-AKT/mTOR | [38] | |
| CV777 | IPEC-J2 | ↓PI3K/Akt ↑mTOR upstream regulators (ATP6V1G1, FZD2 and LAMTOR2), ↑ PTEN | [39] | |
| TGEV Host: swine | Shaanxi | PK-15 | ↓p300/CBP, ↓MDM2 ↑p53, p38 MAPK | [28] |
| Shaanxi | PK-15 | ↑ROS, ↑p53, p38 MAPK | [45] | |
| Shaanxi | PK-15 | ↑AIFM1, PARP cleavage, ↑ROS | [46] | |
| Shaanxi | PK-15 | ↑Fas/FasL, ↑Bid, ↑Bax, ↓Bcl-2 ↑ cleaved PARP, ↑caspase 8,9,3 | [47] | |
| Shaanxi | PK-15 | S and G2/M cycle arrest ↑p53, ↑p21 | [48] | |
| Purdue-11 | HRT18 | caspase 3,6,7,8,9 | [49] | |
| Shaanxi | PK-15 | miR-27b, ↓RUNX1 ↓Bax, caspase 3,9 | [50] | |
| SADS-CoV Host: swine | SADS-CoV | Vero, IPI-2I | ↑ Fas/FasL ↑ caspase 8,9, 3, PARP cleavage | [52] |
| FECV Host: Feline (cat) | 79-1146 | Feline peripheral blood mononuclear cells (PBMC), CD4+ cells, CD8+ cells, CD21+ cells, peritoneal exudate cells (PEC), alveolar macrophages, and WEHI-164 murine sarcoma cells | ↑TNF-α | [56] |
| 79–1146 | CRFK | ↑PDL1 | [57] | |
| FIPV-m3c-2 | Peritoneal cells | ↑ Fas, TNF, Bax, Bak, Bix, Bid, Traf2 | [58] | |
| FIPV 79-1146 | CRFK | ↓TNF, TGFbeta, STAT3 | [59] | |
| FIPV 79-1146 | CRFK | p53, p38 MAPK, VEGF | [60] | |
| CCoV Host: Canine (dog) | Type I, 1–71 | A-72 | ↑ caspase 3 | [63] |
| Type II, S/378 | A-72 | PARP, Bid cleavage ↑ caspase 8,9,3,6 | [64] | |
| Type II, S/378 | A-72 | ↑TRAIL, ↑ Fas/FasL, ↓Bcl-2 | [67] | |
| Betacoronaviruses | ||||
| MHV Host: mouse | A59 | 17Cl-1 | caspase dependent | [78] |
| MHV-3 | A/J and BALB/c macrophages | fgl2 prothrombinase | [79] | |
| MHV-3 | BTLA-deficient (BTLA-/-) mice | TRAIL | [81] | |
| A59 | oligodendrocytes | apoptosis | [84] | |
| JHM | CG-4 | ↑Fas/FasL/FADD/procaspase 8 | [103] | |
| MHV-2 | thymus | apoptosis | [86] | |
| PHEV Host: swine | HEV-67N | PK-15 | ↑ Fas/FasL ↑ caspase 8,9, 3, PARP cleavage | [90] |
| 67Ν | Four- to six-week-old female BALB/c mice | ↑bak1, ↑caspase 1,3,4,7,8,12 | [91] | |
| ECoV Host: horse | NC99 | MDBK | ↑caspase 3,7,8,9 | [94] |
| Gammacoronaviruses | ||||
| IBV Host: avian | Beaudette | Vero | ↑ caspase 3, ↑ cleaved PARP | [99] |
| Beaudette | Vero | p53 independent | [100] | |
| Beaudette | Vero | ↑ Bak | [101] | |
| ck/CH/LDL/091022 | Chicken kidney tissue | ↑Bcl2, Fas, ↓clusterin | [102] | |
| SCDY2 SCK2 | Chicken kidney tissue | BCL2L1, GADD45, STAT3 | [103] | |
| Beaudette | Vero | ↑ PERK, PKR, ATF3, ATF4, GADD153, TRIB3,↓ Bcl2, ERKs | [104] | |
| Beaudette | Vero, H1299, Huh-7 | ↑ MKK7, JNK, ↓Bcl-2 | [105] | |
| Beaudette | Vero, H1299 | ↑ ERK1/2, ↑cJUN, JUNB, JUND, cFOS, FOSB | [106] | |
| Beaudette | Vero | PARP cleavage | [107] | |
| Beaudette | macrophage HD11 | ↑ Fas/FasL,↑ caspase 8,9, 3 ↓Bcl-2 | [108] | |
| Deltacoronaviruses | ||||
| PDCoV Host: swine | KNU16-07 | ST cells | Bax, cytochrome C, caspase 9 | [112] |
| CH/XJYN/2016 | LLC-PK | ↑ caspase 8,7,3, ↓ PARP | [113] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gioti, K.; Kottaridi, C.; Voyiatzaki, C.; Chaniotis, D.; Rampias, T.; Beloukas, A. Animal Coronaviruses Induced Apoptosis. Life 2021, 11, 185. https://doi.org/10.3390/life11030185
Gioti K, Kottaridi C, Voyiatzaki C, Chaniotis D, Rampias T, Beloukas A. Animal Coronaviruses Induced Apoptosis. Life. 2021; 11(3):185. https://doi.org/10.3390/life11030185
Chicago/Turabian StyleGioti, Katerina, Christine Kottaridi, Chrysa Voyiatzaki, Dimitrios Chaniotis, Theodoros Rampias, and Apostolos Beloukas. 2021. "Animal Coronaviruses Induced Apoptosis" Life 11, no. 3: 185. https://doi.org/10.3390/life11030185
APA StyleGioti, K., Kottaridi, C., Voyiatzaki, C., Chaniotis, D., Rampias, T., & Beloukas, A. (2021). Animal Coronaviruses Induced Apoptosis. Life, 11(3), 185. https://doi.org/10.3390/life11030185