
SARS-CoV-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave


 ,
, 
 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Raw Data Pre-Processing and Filtering
2.2. Reconstruction of Phylogenetic Trees and Phylodynamics Analysis
2.3. Identification of Molecular Transmission Clusters (MTCs)
2.4. Statistical Analysis
3. Results
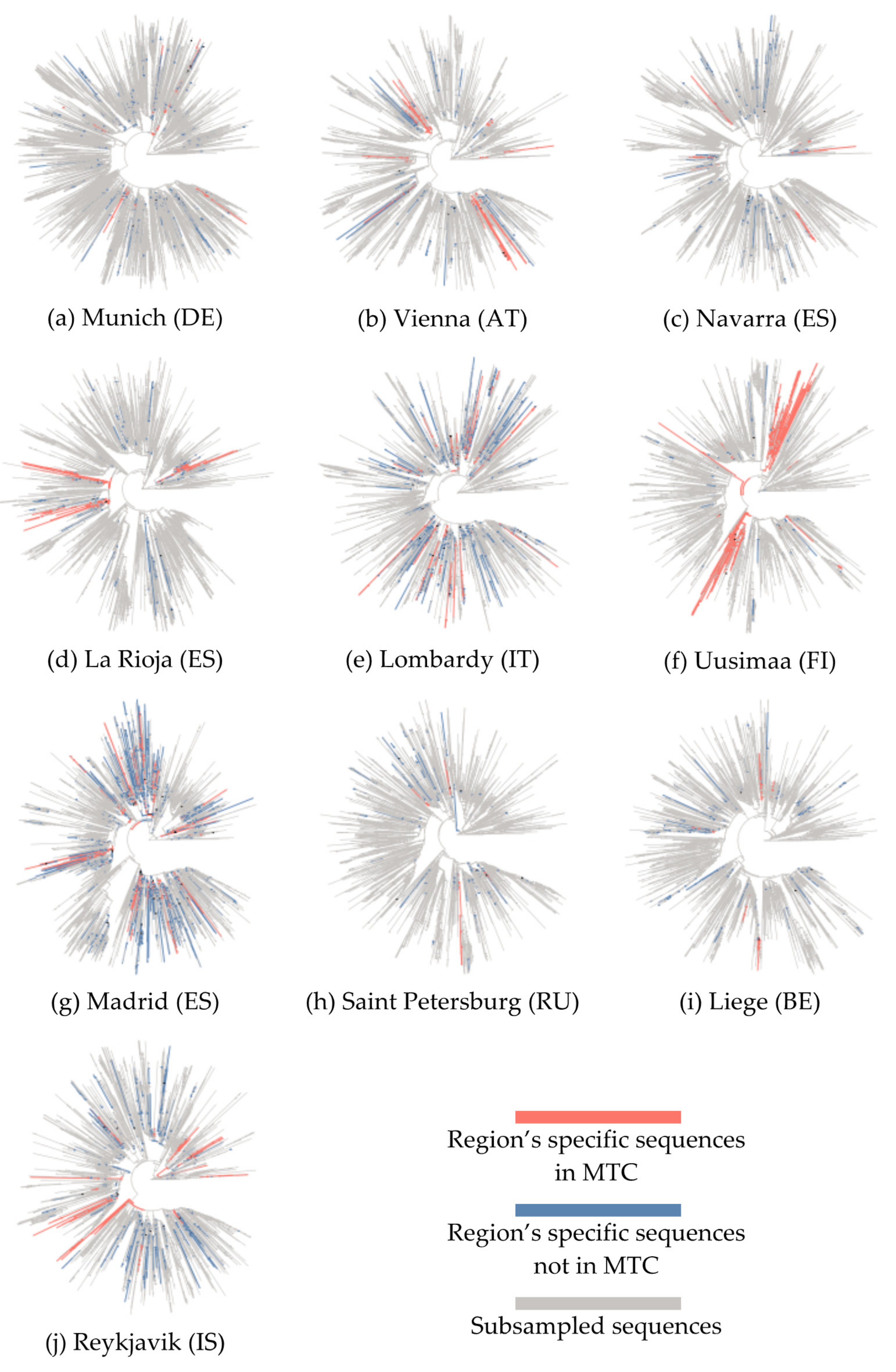
3.1. Phylogenetic Analysis and Characterization of Molecular Transmission Clusters
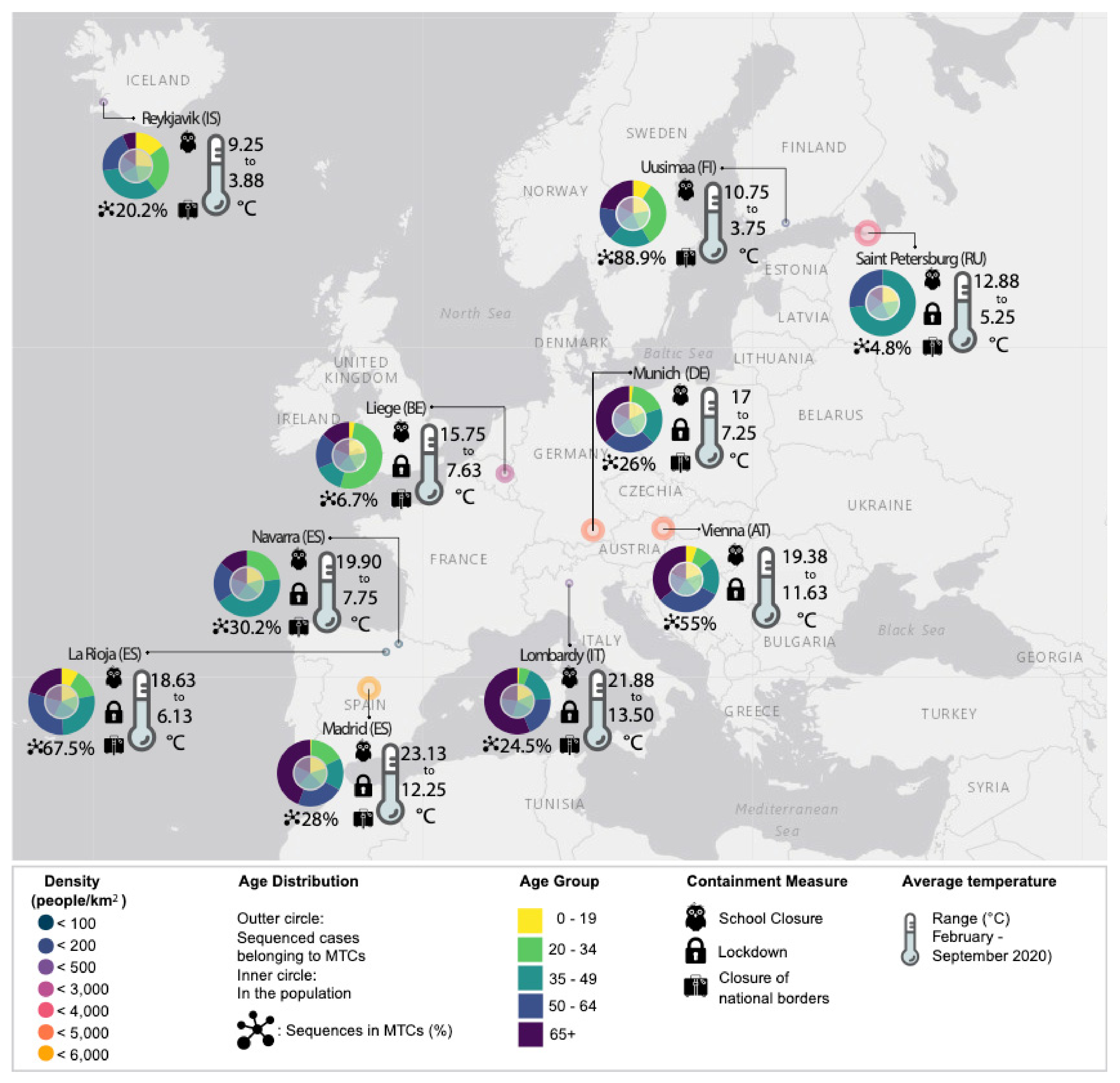
3.2. Demographic Characteristics and Molecular Transmission Clusters
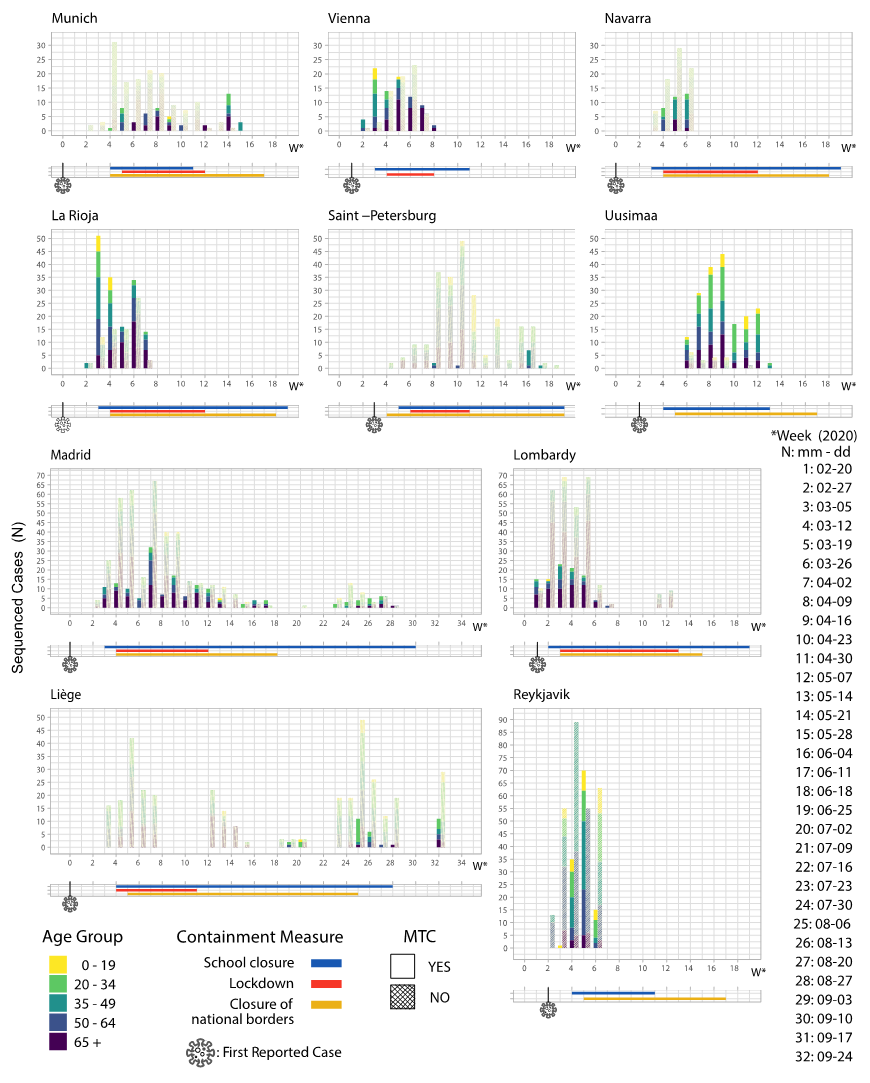
3.3. Containment Measures Taken and Molecular Transmission Clusters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2018, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Drosten, C.; Preiser, W.; Günther, S.; Schmitz, H.; Doerr, H.W. Severe acute respiratory syndrome: Identification of the etiological agent. Trends Mol. Med. 2003, 9, 325–327. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.T.; Lynch, J.B.; del Rio, C. Mild or moderate Covid-19. N. Engl. J. Med. 2020, 383, 1757–1766. [Google Scholar] [CrossRef]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the coronavirus study group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data from vision to reality. Eurosurveillance 2017, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, M.L. Infectious diseases epidemiology. J. Epidemiol. Community Health 2006, 60, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 novel coronavirus (SARS-CoV-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef]
- Petersen, E.; Koopmans, M.; Go, U.; Hamer, D.H.; Petrosillo, N.; Castelli, F.; Storgaard, M.; Al Khalili, S.; Simonsen, L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020, 20, e238–e244. [Google Scholar] [CrossRef]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European region, January to June 2020. Eurosurveillance 2020, 25, 1–8. [Google Scholar] [CrossRef]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, E.F.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. E 2020, 83. [Google Scholar] [CrossRef]
- Bai, Y.; Jiang, D.; Lon, J.R.; Chen, X.; Hu, M.; Lin, S.; Chen, Z.; Wang, X.; Meng, Y.; Du, H.; et al. Comprehensive evolution and molecular characteristics of a large number of SARS-CoV-2 genomes reveal its epidemic trends. Int. J. Infect. Dis. 2020, 100, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.C.F.; Anderson, D.E.; Young, B.E.; Barnaby, E.; Linster, M.; Zhu, F.; Jayakumar, J.; Zhuang, Y.; Kalimuddin, S.L.; Jenny, G.H.; et al. Discovery and genomic characterization of a 382-nucleotide deletion in ORF7B and orf8 during the early evolution of SARS-CoV-2. mBio 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Alinda, A.K.; Ambutsi, M.J.; Kakai, S.K.; Okoth, P.K. SARS-CoV-2 molecular clock and zoonosis. J. Gen. Mol. Virol. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. eLife 2018, 7, e31257. [Google Scholar] [CrossRef] [PubMed]
- Virk, R.K.; Jayakumar, J.; Mendenhall, I.H.; Moorthy, M.; Lam, P.; Linster, M.; Lim, J.; Lin, C.; Oon, L.L.E.; Lee, H.K.; et al. Divergent evolutionary trajectories of influenza B viruses underlie their contemporaneous epidemic activity. Proc. Natl. Acad. Sci. USA 2020, 117, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 evolution and vaccines: Cause for concern? Lancet Respir. Med. 2021, 2600, 19–21. [Google Scholar] [CrossRef]
- Morel, B.; Barbera, P.; Czech, L.; Bettisworth, B.; Hubner, L.; Lutteropp, S.; Serdari, D.; Kostaki, E.G.; Mamais, I.; Kozlov, A.M.; et al. Phylogenetic analysis of SARS-CoV-2 data is difficult. Mol. Biol. Evol. 2020, 1–16. [Google Scholar] [CrossRef]
- Paraskevis, D.; Beloukas, A.; Stasinos, K.; Pantazis, N.; de Mendoza, C.; Bannert, N.; Meyer, L.; Zangerle, R.; Gill, J.; Prins, M.; et al. HIV-1 molecular transmission clusters in nine European countries and Canada: Association with demographic and clinical factors. BMC Med. 2019, 17. [Google Scholar] [CrossRef] [Green Version]
- Rich, S.N.; Richards, V.L.; Mavian, C.N.; Switzer, W.M.; Magalis, B.R.; Poschman, K.; Geary, S.; Broadway, S.E.; Bennett, S.B.; Blanton, J.; et al. Employing molecular phylodynamic methods to identify and forecast HIV transmission clusters in public health settings: A qualitative study. Viruses 2020, 12, 921. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Panneer, N.; France, A.M.; Saduvala, N.; Oster, A.M. Incident infection in high-priority HIV molecular transmission clusters in the United States. AIDS 2020, 34, 1187–1193. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Pond, S.L.K.; Smith, D.M.; Torian, L.V. Growth of HIV-1 Molecular Transmission Clusters in New York City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Abe, E.M.; Li, X.X.; Bergquist, R.; Xu, L.; Xue, J.B.; Ruan, Y.; Cao, C.L.; Li, S.Z. Space-time clustering and associated risk factors of pulmonary tuberculosis in southwest China. Infect. Dis. Poverty 2018, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Oster, A.M.; Switzer, W.M.; Zhang, C.; Panneer, N.; Campbell, E.; Saduvala, N.; Johnson, J.A.; Heneine, W. Natural selection favoring more transmissible HIV detected in United States molecular transmission network. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- di Giallonardo, F.; Pinto, A.N.; Keen, P.; Shaik, A.; Carrera, A.; Salem, H.; Selvey, C.; Nigro, S.J.; Frases, N.; Price, K.; et al. Subtype-specific differences in transmission cluster dynamics of HIV-1 B and CRF01_AE in New South Wales, Australia. J. Int. AIDS Soc. 2019, 24, e25655. [Google Scholar] [CrossRef]
- Kostaki, E.G.; Gova, M.; Adamis, G.; Xylomenos, G.; Chini, M.; Mangafas, M.; Metallidis, S.; Tsachouridou, O.; Papastamopoulos, V.; Chatzidimitriou, D.; et al. A nationwide study about the dispersal patterns of the predominant HIV-1 subtypes A1 and B in Greece: Inference of the molecular transmission clusters. Viruses 2020, 12, 1183. [Google Scholar] [CrossRef]
- Dennis, A.M.; Volz, E.; Frost, S.; Hossain, M.; Poon, A.F.Y.; Rebeiro, P.F.; Vermund, S.H.; Sterling, T.R.; Kalish, M.L. HIV-1 Transmission clustering and phylodynamics highlight the important role of young men who have sex with men. AIDS Res. Hum. Retrovir. 2018, 34, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.; Cowan, S.A.; Nielsen, J.; Fischer, T.K.; Fonager, J. Characterisation of HIV-1 transmission clusters and drug-resistant mutations in Denmark, 2004 to 2016. Eurosurveillance 2018, 23, 44. [Google Scholar] [CrossRef]
- Furuse, Y.; Sando, E.; Tsuchiya, N.; Miyahara, R.; Yasuda, I.; Ko, Y.K.; Saito, M.; Morimoto, K.; Imamura, T.; Shobugawa, Y.; et al. Clusters of coronavirus disease in communities, Japan, January–April 2020. Emerg. Infect. Dis. 2020, 26, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Longini, I.M.; Nizam, A.; Xu, S.; Ungchusak, K.; Hanshaoworakul, W.; Cummings, D.A.T.; Halloran, E.M. Containing pandemic influenza at the source. Science 2005, 309, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Bauch, C.T.; Lloyd-Smith, J.O.; Coffee, M.P.; Galvani, A.P. Dynamically modeling SARS and other newly emerging respiratory illnesses: Past, present, and future. Epidemiology 2005, 16, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Karatayev, V.A.; Anand, M.; Bauch, C.T. Local lockdowns outperform global lockdown on the far side of the COVID-19 epidemic curve. Proc. Natl. Acad. Sci. USA 2020, 117, 24575–24580. [Google Scholar] [CrossRef]
- Koh, W.C.; Naing, L.; Chaw, L.; Rosledzana, M.A.; Alikhan, M.F.; Jamaludin, S.A.; Amin, F.; Omar, A.; Shazli, A.; Griffith, M.; et al. What do we know about SARS-CoV-2 transmission? A systematic review and meta-analysis of the secondary attack rate and associated risk factors. PLoS ONE 2020, 15, e0240205. [Google Scholar] [CrossRef]
- Li, C.; Romagnani, P.; von Brunn, A.; Anders, H.-J. SARS-CoV-2 and Europe: Timing of containment measures for outbreak control. Infection 2020, 48, 483–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primc, K.; Slabe-Erker, R. The success of public health measures in Europe during the COVID-19 pandemic. Sustainability 2020, 12, 4321. [Google Scholar] [CrossRef]
- Meier, K.; Glatz, T.; Guijt, M.C.; Piccininni, M.; van der Meulen, M.; Atmar, K.; Jolink, A.-T.C.; Kurth, T.; Rohmann, J.L.; Najafabadi, A.H.Z. Public perspectives on protective measures during the COVID-19 pandemic in the Netherlands, Germany and Italy: A survey study. PLoS ONE 2020, 15, e0236917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Litvinova, M.; Liang, Y.; Wang, Y.; Wang, W.; Zhao, S.; Wu, Q.; Merler, S.; Viboud, C.; Vespignani, A.; et al. Changes in contact patterns shape the dynamics of the COVID-19 outbreak in China. Science 2020, 368, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- de Salazar, P.M.; Gomez-Barroso, D.; Pampaka, D.; Gill, J.M.; Penalver, B.; Fernandez-Escobar, C.; Lipsitch, M.; Larrauri, A.; Goldstein, E.; Hernan, M.A. Lockdown measures and relative changes in the age-specific incidence of SARS-CoV-2 in Spain. Epidemiol. Infect. 2020, 148, e268. [Google Scholar] [CrossRef]
- Sannigrahi, S.; Pilla, F.; Basu, B.; Basu, A.S.; Molter, A. Examining the association between socio-demographic composition and COVID-19 fatalities in the European region using spatial regression approach. Sustain. Cities Soc. 2020, 62, 102418. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009, 537, 39–64. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. Model finder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turakhia, Y.; De Maio, N.; Thornlow, B.; Gozashti, L.; Lanfear, R.; Walker, C.R.; Hinrichs, A.S.; Fernandes, J.D.; Borges, R.; Slodkowicz, G.; et al. Stability of SARS-CoV-2 phylogenies. PLoS Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pipes, L.; Wang, H.; Huelsenbeck, J.P.; Nielsen, R. Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Jones, L.R.; Manrique, J.M. Assessing SARS-CoV-2 spatial phylogenetic structure: Evidence from RNA and protein sequences. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, T.; Liu, D.; Yang, Y.; Guo, J.; Feng, Y.; Zhang, X.; Cheng, S.; Feng, J. Phylogenetic supertree reveals detailed evolution of SARS-CoV-2. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chalmet, K.; Staelens, D.; Blot, S.; Dinakis, S.; Pelgrom, J.; Plum, J.; Vogalaers, D.; Vandekerckhove, L.; Verhofstede, C. Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect. Dis. 2010, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraskevis, D.; Nikolopoulos, G.; Sypsa, V.; Psichigiou, M.; Pantavou, K.; Kostaki, E.; Karamitros, T.; Paraskeva, D.; Schneider, J.; Malliori, M.; et al. Molecular investigation of HIV-1 cross-group transmissions during an outbreak among people who inject drugs (2011–2014) in Athens, Greece. Infect. Genet. Evol. 2018, 62, 11–16. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69. [Google Scholar] [CrossRef] [PubMed]
- Pollán, M.; Pérez-Gómez, B.; Pastor-Barriuso, R.; Oteo, J.; Hernán, M.A.; Pérez-Olmeda, M.; Sanmartín, J.L.; Fernández-García, A.; Cruz, I.; de Larrea, N.F.; et al. Prevalence of SARS-CoV-2 in Spain (ENE-COVID): A nationwide, population-based seroepidemiological study. Lancet 2020, 396, 535–544. [Google Scholar] [CrossRef]
- Lemieux, J.E.; Siddle, K.J.; Shaw, B.M.; Loreth, C.; Schaffner, S.F.; Gladden-Young, A.; Adams, G.; Fink, T.; Tomkins-Tinch, C.H.; Krasilnikova, L.A.; et al. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Am. Assoc. Adv. Sci. 2021, 371. [Google Scholar] [CrossRef]
- Goldstein, E.; Lipsitch, M. Temporal rise in the proportion of younger adults and older adolescents among coronavirus disease (COVID-19) cases following the introduction of physical distancing measures, Germany, March to April 2020. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef]
- Singh, S.; Roy, D.; Sinha, K.; Parveen, S.; Sharma, G.; Joshi, G. Impact of COVID-19 and lockdown on mental health of children and adolescents: A narrative review with recommendations. Psychiatry Res. 2020, 293, 113429. [Google Scholar] [CrossRef]
- Pierce, M.; Hope, H.; Ford, T.; Hatch, S.; Hotopf, M.; John, A.; Kontopantelis, E.; Webb, R.; Wessely, S.; McManus, S.; et al. Mental health before and during the COVID-19 pandemic: A longitudinal probability sample survey of the UK population. Lancet Psychiatry 2020, 7, 883–892. [Google Scholar] [CrossRef]
- Townsend, E. COVID-19 policies in the UK and consequences for mental health. Lancet Psychiatry 2020, 7, 1014–1015. [Google Scholar] [CrossRef]
- Leclerc, Q.J.; Fuller, N.M.; Knight, L.E.; Funk, S.; Knight, G.M. What settings have been linked to SARS-CoV-2 transmission clusters? Wellcome Open Res. 2020, 5. [Google Scholar] [CrossRef]
- Adam, D.C.; Wu, P.; Wong, J.Y.; Lau, E.H.; Tsang, T.K.; Cauchemez, S.; Leung, G.M.; Cowling, B.J. Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong. Nat. Med. 2020, 26, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Viner, R.M.; Russell, S.J.; Croker, H.; Packer, J.; Ward, J.; Stansfield, C.; Mytton, O.; Bonell, C.; Booy, R. School closure and management practices during coronavirus outbreaks including COVID-19: A rapid systematic review. Lancet Child Adolesc. Health 2020, 4, 397–404. [Google Scholar] [CrossRef]
- Götzinger, F.; Santiago-García, B.; Noguera-Julián, A.; Lanaspa, M.; Lancella, L.; Carducci, F.I.; Gabrovska, N.; Velizarova, S.; Prunk, P.; Osterman, V.; et al. COVID-19 in children and adolescents in Europe: A multinational, multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 653–661. [Google Scholar] [CrossRef]
- Esposito, S.; Principi, N. Debates Around the Role of School Closures in the Coronavirus 2019 Pandemic-Reply. J. Assoc. Med. Am. Pediatr. 2020. [Google Scholar] [CrossRef]
- Cao, Q.; Chen, Y.-C.; Chen, C.-L.; Chiu, C.-H. SARS-CoV-2 infection in children: Transmission dynamics and clinical characteristics. J. Formos. Med. Assoc. 2020, 119, 670–673. [Google Scholar] [CrossRef]
- Piovani, D.; Christodoulou, M.N.; Hadjidemetriou, A.; Pantavou, K.; Zaza, P.; Bagos, P.G.; Bonovas, S.; Nikolopoulos, G.K. Effect of early application of social distancing interventions on COVID-19 mortality over the first pandemic wave: An analysis of longitudinal data from 37 countries. J. Infect. 2020. [Google Scholar] [CrossRef]
- Wells, C.R.; Sah, P.; Moghadas, S.M.; Pandey, A.; Shoukat, A.; Wang, Y.; Wang, Z.; Meyers, L.A.; Singer, B.H.; Galvani, A.P. Impact of international travel and border control measures on the global spread of the novel 2019 coronavirus outbreak. Proc. Natl. Acad. Sci. USA 2020, 117, 7504–7509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St. John, R.K.; King, A.; de Jong, D.; Bodie-Collins, M.; Squires, S.G.; Tam, T.W.S. Border screening for SARS. Emerg. Infect. Dis. 2005, 11, 6–10. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Kee, T.G.; Paton, N.I. Experience of severe acute respiratory syndrome in Singapore: Importation of cases, and defense strategies at the airport. J. Travel Med. 2006, 10, 259–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| EU Geographical Region | Region’s Specific Sequences, N | Total Sequences in Tree, N | Samples Collection Period | MTCs, N | Sequences in MTCs, N (%) | Number of Sequences in Each MTC, N | Region’s Specific Sequences Enrichment, N (%) | MTC Clades |
|---|---|---|---|---|---|---|---|---|
| Munich (DE) | 195 | 8032 | 2020-03-02 to 2020-05-26 | 5 | 51 (26.0) | 28 17 12 6 5 | 19 (67.9) 16 (94.1) 8 (66.7) 4 (66.7) 4 (80.0) | G 32/51 GR 19/51 |
| Vienna (AT) | 149 | 2990 | 2020-02-26 to 2020-04-14 | 8 | 82 (55.0) | 44 10 9 9 8 6 6 5 | 38 (86.4) 9 (90.0) 8 (88.9) 8 (88.9) 6 (75.0) 5 (83.3) 4 (66.7) 4 (80.0) | G 9/82 GR 59/82 S 14/82 |
| Navarra (ES) | 109 | 3646 | 2020-03-07 to 2020-03-29 | 3 | 33 (30.2) | 24 11 7 | 17 (70.8) 11 (100.0) 5 (71.4) | GR 17/33 S 16/33 |
| La Rioja (ES) | 256 | 3779 | 2020-02-29 to 2020-04-04 | 9 | 173 (67.5) | 68 51 22 21 16 9 8 6 6 | 62 (91.2) 35 (68.6) 20 (90.9) 19 (90.5) 15 (93.7) 8 (88.9) 6 (75.0) 4 (66.7) 4 (66.7) | GH 74/173 S 99/173 |
| Lombardy (IT) | 412 | 3333 | 2020-02-20 to 2020-05-10 | 14 | 101 (24.5) | 16 12 12 12 11 9 8 8 7 6 6 5 5 5 | 12 (75.0) 12 (100.0) 11 (91.7) 8 (66.7) 8 (72.7) 8 (88.9) 7 (87.5) 6 (75.0) 6 (85.7) 5 (83.3) 5 (83.3) 5 (100.0) 4 (80.0) 4 (80.0) | G 81/101 GR 20/101 |
| Uusimaa (FI) | 227 | 2979 | 2020-03-13 to 2020-05-16 | 7 | 202 (88.9) | 156 66 40 6 6 6 6 5 | 109 (69.9) 44 (66.7) 31 (77.5) 5 (83.3) 5 (83.3) 4 (66.7) 4 (66.7) 4 (80.0) | G 17/202 GH 185/202 |
| Madrid (ES) | 582 | 4025 | 2020-02-25 to 2020-08-30 | 20 | 163 (28.0) | 26 20 19 18 16 14 13 13 9 8 7 6 6 6 6 5 5 5 5 5 | 17 (65.4) 14 (70.0) 13 (68.4) 13 (72.2) 13 (81.2) 13 (92.9) 11 (84.6) 10 (76.9) 8 (88.9) 7 (87.5) 6 (85.7) 5 (83.3) 5 (83.3) 4 (66.7) 4 (66.7) 4 (80.0) 4 (80.0) 4 (80.0) 4 (80.0) 4 (80.0) | G 108/163 S 50/163 V 5/163 |
| Saint Petersburg (RU) | 267 | 2967 | 2020-03-13 to 2020-06-16 | 2 | 13 (4.8) | 11 5 | 9 (81.8) 4 (80.0) | G 4/13 GR 9/13 |
| Liege (BE) | 535 | 3349 | 2020-03-05 to 2020-09-25 | 3 | 36 (6.7) | 27 10 6 | 21 (77.8) 10 (100.0) 5 (83.3) | G 10/36 GR 26/36 |
| Reykjavik (IS) | 601 | 3044 | 2020-02-27 to 2020-03-29 | 6 | 122 (20.2) | 62 47 14 12 9 7 | 55 (88.7) 31 (66.0) 12 (85.7) 12 (100.0) 6 (66.7) 6 (85.7) | G 62/122 GR 12/122 L 6/122 O 2/122 S 6/122 V 34/122 |
| Munich (DE) | Vienna (AT) | Navarra (ES) | La Rioja (ES) | Lombardy (IT) | Uusimaa (FI) | Madrid (ES) | Saint Petersburg (RU) | Liege (BE) | Reykjavik (IS) | Total CL/UN | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clustered/Unclustered sequences | ||||||||||||
| AGE | 0–19 | 1/4 | 5/4 | 0/1 | 11/3 | 1/3 | 17/3 | 1/8 | 0/23 | 1/15 | 18/33 | 68/133 |
| 20–34 | 9/28 | 8/7 | 8/6 | 18/7 | 5/10 | 60/4 | 27/60 | 0/44 | 18/95 | 29/113 | 194/405 | |
| 35–49 | 9/26 | 17/24 | 15/21 | 36/20 | 17/39 | 38/2 | 25/70 | 8/49 | 5/87 | 41/157 | 233/594 | |
| 50–64 | 13/37 | 29/7 | 7/26 | 40/21 | 19/57 | 30/4 | 36/110 | 3/74 | 6/70 | 25/131 | 243/615 | |
| 65–100 | 19/48 | 33/24 | 5/20 | 27/33 | 54/184 | 41/5 | 70/170 | 0/47 | 5/79 | 8/21 | 295/693 | |
| Overall | 51/143 | 92/66 | 35/74 | 13/74 | 96/293 | 186/18 | 159/418 | 11/237 | 35/346 | 121/455 | 1033/2440 | |
| Total | 194 | 158 | 109 | 206 | 389 | 204 | 577 | 248 | 381 | 576 | 3473 | |
| Chi’s p-value | NS | NS | NS | NS | NS | NS | NS | ** | * | * | NS | |
| GENDER | Female | 25/54 | 42/26 | 21/41 | 65/29 | 40/132 | 90/9 | 84/206 | 6/143 | 11/151 | 61/214 | 503/1144 |
| Male | 26/90 | 40/40 | 12/34 | 61/35 | 56/162 | 95/9 | 79/213 | 7/98 | 19/163 | 60/241 | 512/1262 | |
| Overall | 51/144 | 82/66 | 33/75 | 126/64 | 96/294 | 185/18 | 163/419 | 13/241 | 30/314 | 121/455 | 1015/2406 | |
| Total | 195 | 148 | 108 | 190 | 390 | 203 | 582 | 254 | 344 | 576 | 3421 | |
| Chi’s p-value | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bousali, M.; Dimadi, A.; Kostaki, E.-G.; Tsiodras, S.; Nikolopoulos, G.K.; Sgouras, D.N.; Magiorkinis, G.; Papatheodoridis, G.; Pogka, V.; Lourida, G.; et al. SARS-CoV-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave. Life 2021, 11, 219. https://doi.org/10.3390/life11030219
Bousali M, Dimadi A, Kostaki E-G, Tsiodras S, Nikolopoulos GK, Sgouras DN, Magiorkinis G, Papatheodoridis G, Pogka V, Lourida G, et al. SARS-CoV-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave. Life. 2021; 11(3):219. https://doi.org/10.3390/life11030219
Chicago/Turabian StyleBousali, Maria, Aristea Dimadi, Evangelia-Georgia Kostaki, Sotirios Tsiodras, Georgios K. Nikolopoulos, Dionyssios N. Sgouras, Gkikas Magiorkinis, George Papatheodoridis, Vasiliki Pogka, Giota Lourida, and et al. 2021. "SARS-CoV-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave" Life 11, no. 3: 219. https://doi.org/10.3390/life11030219
APA StyleBousali, M., Dimadi, A., Kostaki, E. -G., Tsiodras, S., Nikolopoulos, G. K., Sgouras, D. N., Magiorkinis, G., Papatheodoridis, G., Pogka, V., Lourida, G., Argyraki, A., Angelakis, E., Sourvinos, G., Beloukas, A., Paraskevis, D., & Karamitros, T. (2021). SARS-CoV-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave. Life, 11(3), 219. https://doi.org/10.3390/life11030219