
Oligodendroglial Energy Metabolism and (re)Myelination


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oligodendroglial Functions Require Energy and Biosynthetic Precursors
2.1. Energetic Cost of Myelin Synthesis
2.1.1. Lipid Synthesis
2.1.2. Protein Synthesis
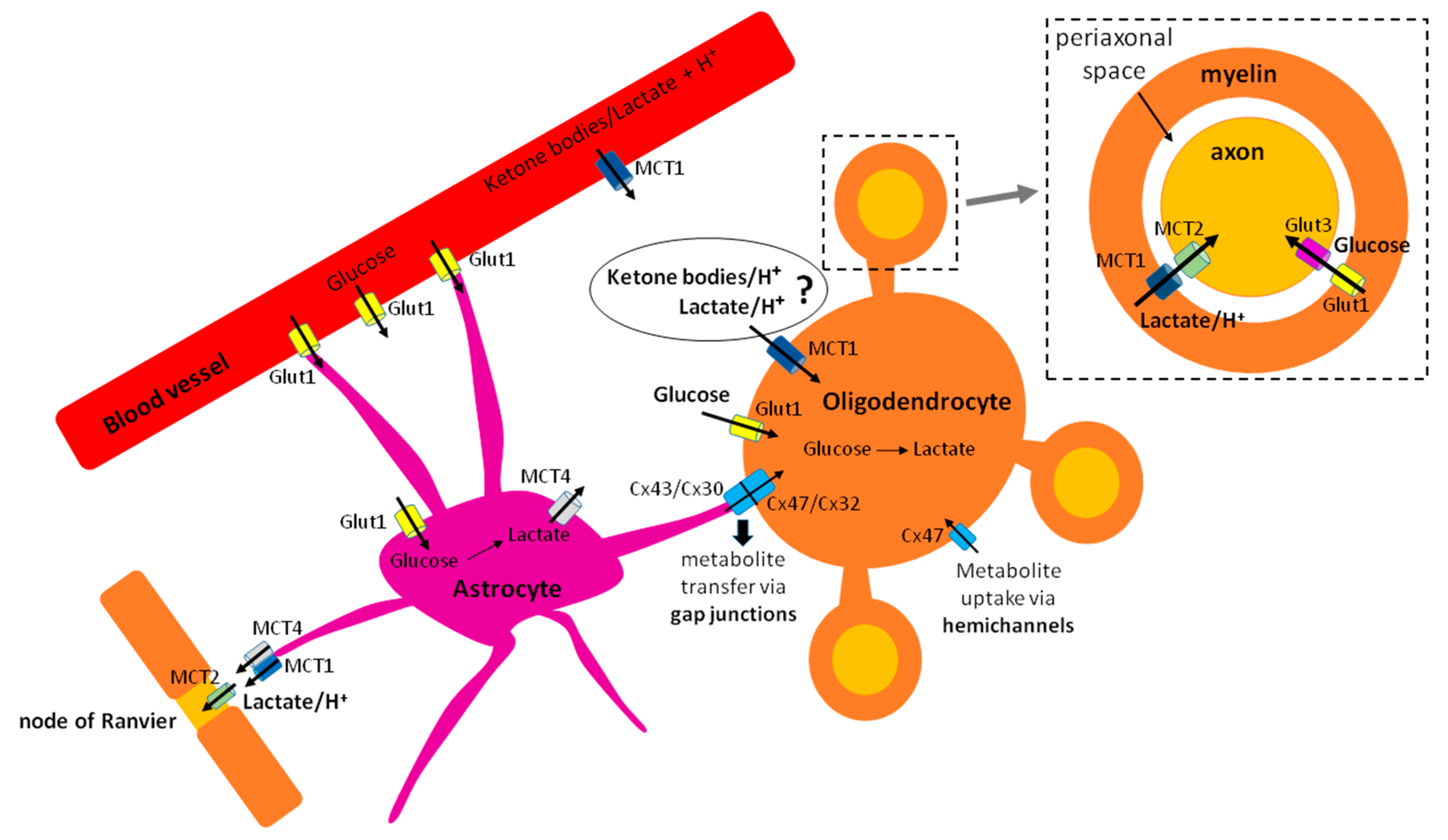
2.2. Oligodendroglial Trophic Support to the Axons Requieres Energy Investment and/or Metabolite Shuttling
3. Energy Fuels Used by Oligodendroglia
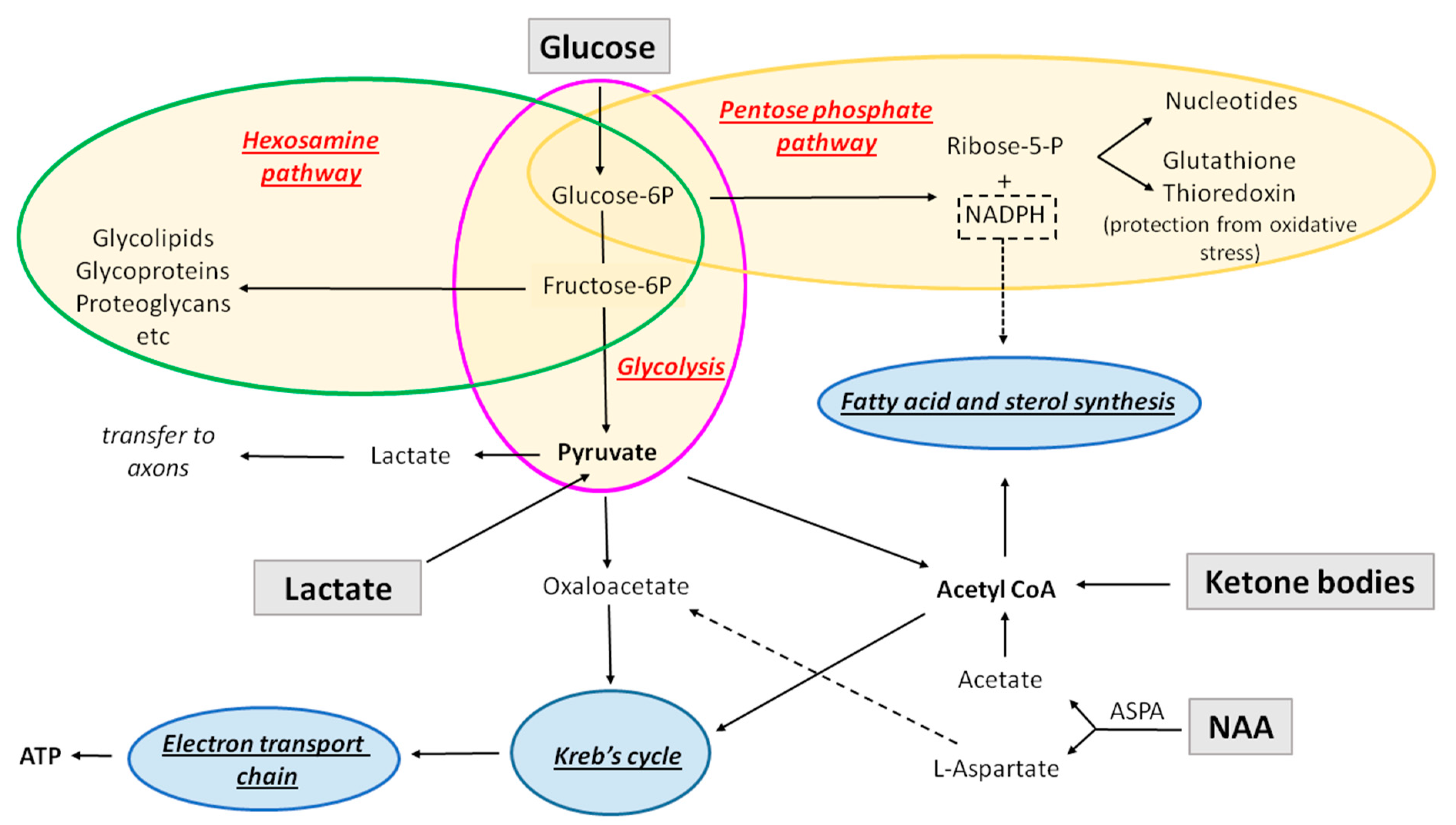
3.1. Glucose
3.1.1. Metabolizing Pathways
3.1.2. Glucose Metabolism in Oligodendroglia: Variations across the Lineage
3.1.3. Glycolysis versus Mitochondrial Metabolism
3.1.4. Pentose Phosphate Pathway
3.1.5. Hexosamine Pathway
4. Alternative Sources of Energy and Biosynthetic Precursors
4.1. Monocarboxylates (Ketone Bodies and Lactate) and Their Transporters in Oligodendroglia
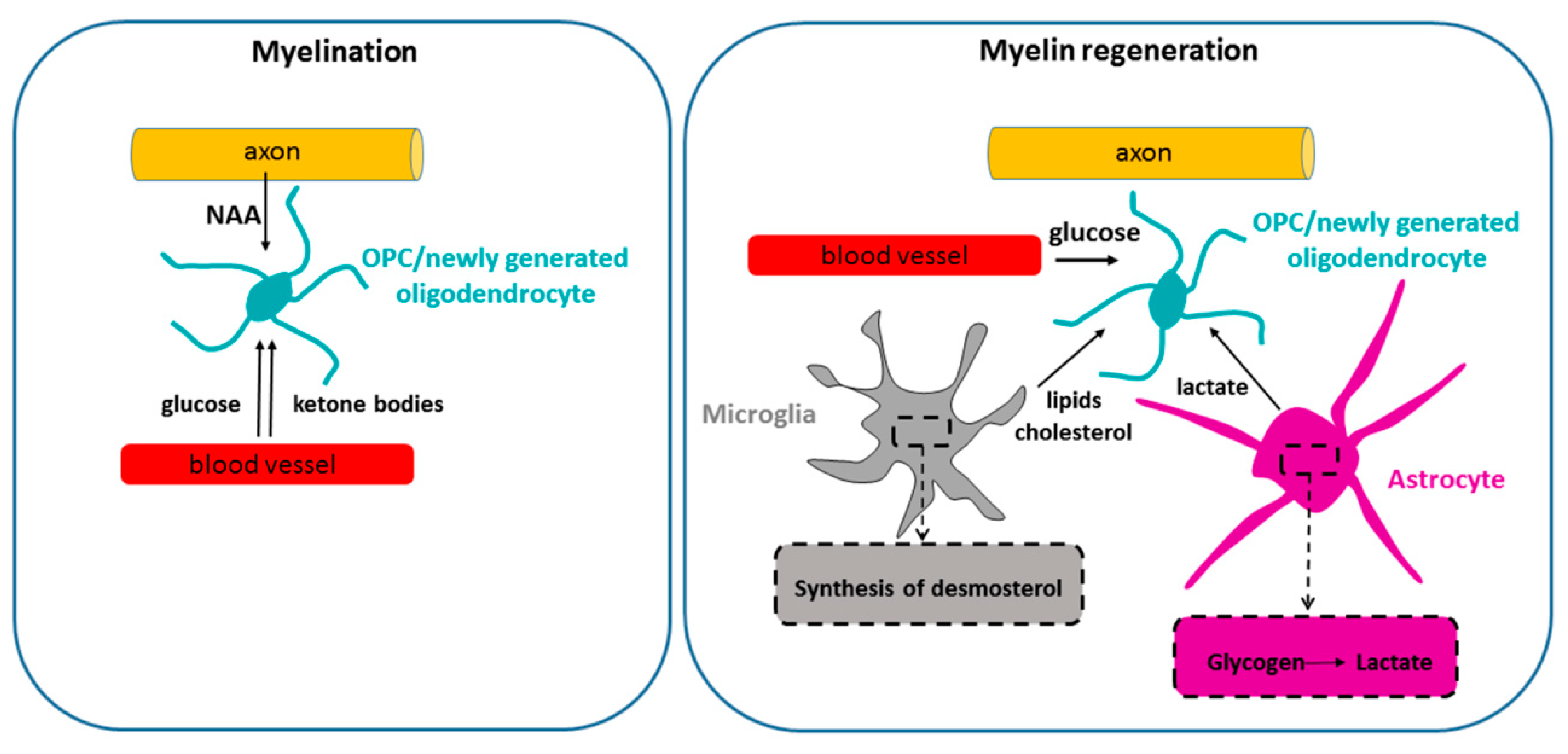
4.1.1. Ketone Bodies as an Important Energy Fuel during Developmental Myelination

4.1.2. Lactate Metabolism in Oligodendroglia: Differentiation State Matters?
4.2. N-Acetyl-Aspartate Sustains Developmental Myelination
5. Energetic Support of Remyelination?
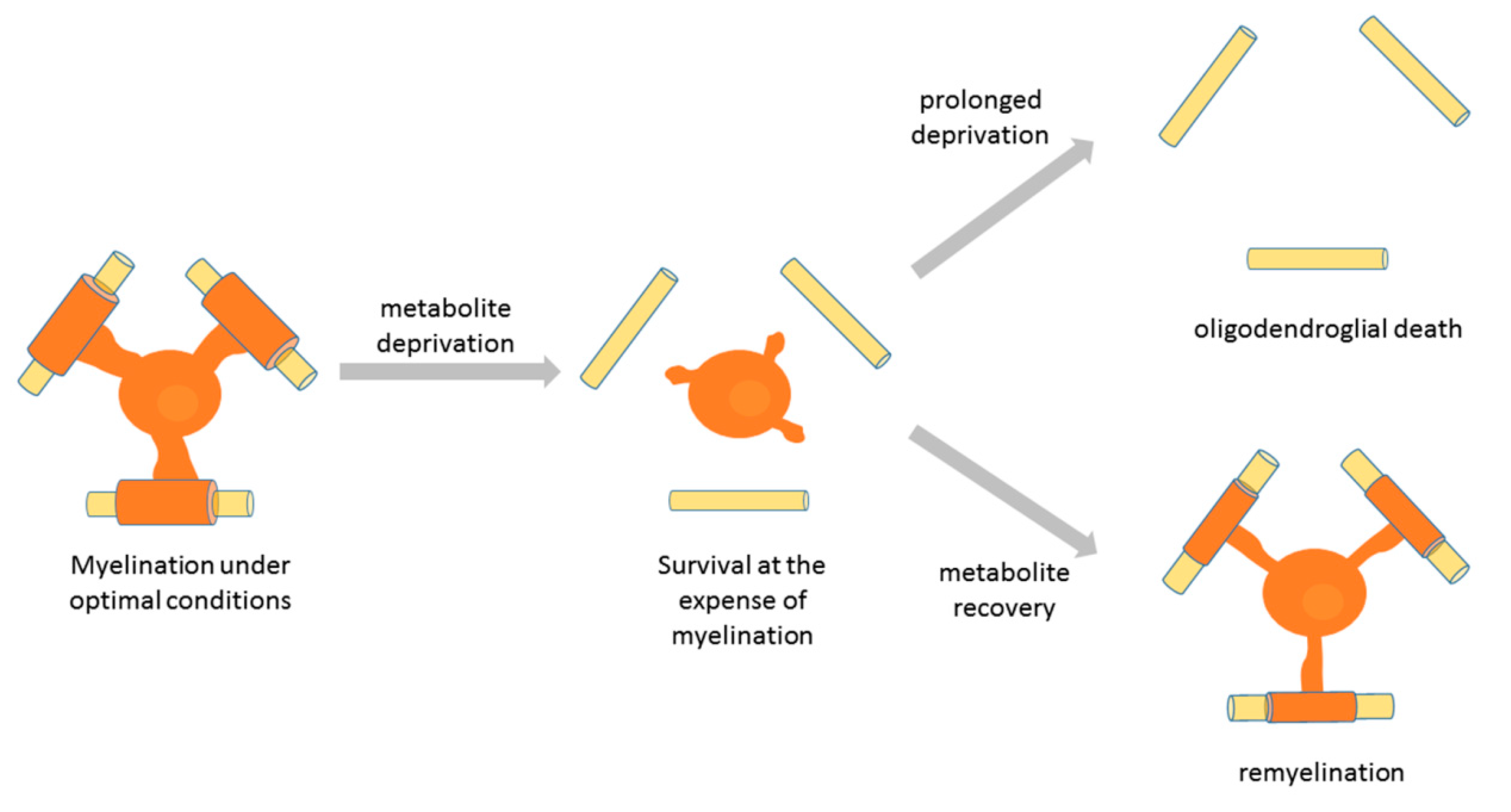
6. Distinct Differentiation Stage-Specific Responses of Oligodendroglia to Nutrient Deprivation: Relevance to Demyelinating Pathology
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zalc, B. The acquisition of myelin: A success story. Novartis Found. Symp. 2006, 276, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.A.; Werner, H.B. Myelination of the nervous system: Mechanisms and functions. Annu Rev. Cell Dev. Biol 2014, 30, 503–533. [Google Scholar] [CrossRef]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131 Pt 6, 1464–1477. [Google Scholar] [CrossRef] [Green Version]
- Edgar, J.M.; McLaughlin, M.; Werner, H.B.; McCulloch, M.C.; Barrie, J.A.; Brown, A.; Faichney, A.B.; Snaidero, N.; Nave, K.A.; Griffiths, I.R. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia 2009, 57, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Edgar, J.M.; McLaughlin, M.; Yool, D.; Zhang, S.C.; Fowler, J.H.; Montague, P.; Barrie, J.A.; McCulloch, M.C.; Duncan, I.D.; Garbern, J.; et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 2004, 166, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Chrast, R.; Saher, G.; Nave, K.A.; Verheijen, M.H. Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. J. Lipid Res. 2011, 52, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T. Therapeutic Strategies for Oligodendrocyte-Mediated Remyelination. Adv. Exp. Med. Biol. 2019, 1190, 265–279. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B. Inherited and acquired disorders of myelin: The underlying myelin pathology. Exp. Neurol. 2016, 283, 452–475. [Google Scholar] [CrossRef]
- Boshans, L.L.; Sherafat, A.; Nishiyama, A. The effects of developmental and current niches on oligodendrocyte precursor dynamics and fate. Neurosci. Lett. 2020, 715, 134593. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Crawford, A.H.; Stockley, J.H.; Tripathi, R.B.; Richardson, W.D.; Franklin, R.J. Oligodendrocyte progenitors: Adult stem cells of the central nervous system? Exp. Neurol. 2014, 260, 50–55. [Google Scholar] [CrossRef]
- Young, K.M.; Psachoulia, K.; Tripathi, R.B.; Dunn, S.J.; Cossell, L.; Attwell, D.; Tohyama, K.; Richardson, W.D. Oligodendrocyte dynamics in the healthy adult CNS: Evidence for myelin remodeling. Neuron 2013, 77, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Gibson, E.M.; Geraghty, A.C.; Monje, M. Bad wrap: Myelin and myelin plasticity in health and disease. Dev. Neurobiol. 2018, 78, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, A.; Mangin, J.M.; Aguirre, A.; Gallo, V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat. Neurosci. 2010, 13, 287–289. [Google Scholar] [CrossRef] [Green Version]
- Sahel, A.; Ortiz, F.C.; Kerninon, C.; Maldonado, P.P.; Angulo, M.C.; Nait-Oumesmar, B. Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front. Cell Neurosci. 2015, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, F.C.; Habermacher, C.; Graciarena, M.; Houry, P.Y.; Nishiyama, A.; Nait Oumesmar, B.; Angulo, M.C. Neuronal activity in vivo enhances functional myelin repair. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTigue, D.M.; Tripathi, R.B. The life, death, and replacement of oligodendrocytes in the adult CNS. J. Neurochem. 2008, 107, 1–19. [Google Scholar] [CrossRef]
- Zhou, X.; He, C.; Ren, J.; Dai, C.; Stevens, S.R.; Wang, Q.; Zamler, D.; Shingu, T.; Yuan, L.; Chandregowda, C.R.; et al. Mature myelin maintenance requires Qki to coactivate PPARβ-RXRα-mediated lipid metabolism. J. Clin. Investig. 2020, 130, 2220–2236. [Google Scholar] [CrossRef] [Green Version]
- Ando, S.; Tanaka, Y.; Toyoda, Y.; Kon, K. Turnover of myelin lipids in aging brain. Neurochem. Res. 2003, 28, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Meschkat, M.; Steyer, A.M.; Weil, M.-T.; Kusch, K.; Jahn, O.; Piepkorn, L.; Agüi-Gonzalez, P.; Ngoc Phan, N.T.; Ruhwedel, T.; Sadowski, B.; et al. White matter integrity requires continuous myelin synthesis at the inner tongue. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’ disease. Sci. Transl. Med. 2010, 2, 52–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, M.S.; Brewer, G.J. Mitoenergetic failure in Alzheimer disease. Am. J. Physiol. Cell Physiol. 2007, 292, C8–C23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Mochel, F.; Haller, R.G. Energy deficit in Huntington disease: Why it matters. J. Clin. Investig. 2011, 121, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Petzold, A.; Tavazzi, B.; Eikelenboom, J.; Keir, G.; Belli, A.; Giovannoni, G.; Di Pietro, V.; Polman, C.; D’Urso, S.; et al. Increase of uric acid and purine compounds in biological fluids of multiple sclerosis patients. Clin. Biochem. 2009, 42, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Amorini, A.M.; Nociti, V.; Petzold, A.; Gasperini, C.; Quartuccio, E.; Lazzarino, G.; Di Pietro, V.; Belli, A.; Signoretti, S.; Vagnozzi, R.; et al. Serum lactate as a novel potential biomarker in multiple sclerosis. Biochim. Biophys. Acta 2014, 1842, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Lazzarino, G.; Amorini, A.M.; Eikelenboom, M.J.; Killestein, J.; Belli, A.; Di Pietro, V.; Tavazzi, B.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.; et al. Cerebrospinal fluid ATP metabolites in multiple sclerosis. Mult. Scler. 2010, 16, 549–554. [Google Scholar] [CrossRef]
- Lazzarino, G.; Amorini, A.M.; Petzold, A.; Gasperini, C.; Ruggieri, S.; Quartuccio, M.E.; Di Stasio, E.; Tavazzi, B. Serum Compounds of Energy Metabolism Impairment Are Related to Disability, Disease Course and Neuroimaging in Multiple Sclerosis. Mol. Neurobiol. 2017, 54, 7520–7533. [Google Scholar] [CrossRef]
- Petzold, A.; Nijland, P.G.; Balk, L.J.; Amorini, A.M.; Lazzarino, G.; Wattjes, M.P.; Gasperini, C.; van der Valk, P.; Tavazzi, B.; van Horssen, J. Visual pathway neurodegeneration winged by mitochondrial dysfunction. Ann. Clin. Transl. Neurol. 2015, 2, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; van der Pol, S.M.; van Het Hof, B.; Reijerkerk, A.; Pellerin, L.; van der Valk, P.; de Vries, H.E.; et al. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Molenaar, R.J.; van der Pol, S.M.; van der Valk, P.; van Noorden, C.J.; de Vries, H.E.; van Horssen, J. Differential expression of glucose-metabolizing enzymes in multiple sclerosis lesions. Acta Neuropathol. Commun. 2015, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef]
- Witte, M.E.; Bø, L.; Rodenburg, R.J.; Belien, J.A.; Musters, R.; Hazes, T.; Wintjes, L.T.; Smeitink, J.A.; Geurts, J.J.; De Vries, H.E.; et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J. Pathol. 2009, 219, 193–204. [Google Scholar] [CrossRef]
- Montani, L. Lipids in regulating oligodendrocyte structure and function. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Brügger, B.; Lappe-Siefke, C.; Möbius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef]
- Saher, G.; Simons, M. Cholesterol and myelin biogenesis. Subcell Biochem. 2010, 51, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef] [PubMed]
- Dimas, P.; Montani, L.; Pereira, J.A.; Moreno, D.; Trötzmüller, M.; Gerber, J.; Semenkovich, C.F.; Köfeler, H.C.; Suter, U. CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife 2019, 8. [Google Scholar] [CrossRef]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Björkhem, I. Crossing the barrier: Oxysterols as cholesterol transporters and metabolic modulators in the brain. J. Intern. Med. 2006, 260, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Kalwy, S.A.; Smith, R. Mechanisms of myelin basic protein and proteolipid protein targeting in oligodendrocytes (review). Mol. Membr. Biol. 1994, 11, 67–78. [Google Scholar] [CrossRef]
- Müller, C.; Bauer, N.M.; Schäfer, I.; White, R. Making myelin basic protein -from mRNA transport to localized translation. Front. Cell Neurosci. 2013, 7, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, W.; Ozgen, H.; Klunder, B.; de Jonge, J.C.; Nomden, A.; Plat, A.; Trifilieff, E.; de Vries, H.; Hoekstra, D. The major myelin-resident protein PLP is transported to myelin membranes via a transcytotic mechanism: Involvement of sulfatide. Mol. Cell Biol. 2015, 35, 288–302. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Rinholm, J.E.; Hamilton, N.B.; Kessaris, N.; Richardson, W.D.; Bergersen, L.H.; Attwell, D. Regulation of oligodendrocyte development and myelination by glucose and lactate. J. Neurosci. 2011, 31, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef] [Green Version]
- Philippot, C.; Griemsmann, S.; Jabs, R.; Seifert, G.; Kettenmann, H.; Steinhäuser, C. Astrocytes and oligodendrocytes in the thalamus jointly maintain synaptic activity by supplying metabolites. Cell Rep. 2021, 34, 108642. [Google Scholar] [CrossRef]
- Kassmann, C.M. Myelin peroxisomes—Essential organelles for the maintenance of white matter in the nervous system. Biochimie 2014, 98, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Frühbeis, C.; Kuo-Elsner, W.P.; Müller, C.; Barth, K.; Peris, L.; Tenzer, S.; Möbius, W.; Werner, H.B.; Nave, K.A.; Fröhlich, D.; et al. Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol. 2020, 18, e3000621. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custódio, T.F.; Paulsen, P.A.; Frain, K.M.; Pedersen, B.P. Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef]
- Arluison, M.; Quignon, M.; Nguyen, P.; Thorens, B.; Leloup, C.; Penicaud, L. Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain—An immunohistochemical study. J. Chem. Neuroanat. 2004, 28, 117–136. [Google Scholar] [CrossRef]
- Arluison, M.; Quignon, M.; Thorens, B.; Leloup, C.; Penicaud, L. Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J. Chem. Neuroanat. 2004, 28, 137–146. [Google Scholar] [CrossRef]
- Zalc, B.; Fields, R.D. Do Action Potentials Regulate Myelination? Neuroscientist 2000, 6, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Matute, C.; Palma, A.; Serrano-Regal, M.P.; Maudes, E.; Barman, S.; Sánchez-Gómez, M.V.; Domercq, M.; Goebels, N.; Dalmau, J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann. Neurol. 2020, 87, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.I.; Hadera, M.G.; Tavares, J.M.; Kotter, M.R.; Sonnewald, U. Characterization of glucose-related metabolic pathways in differentiated rat oligodendrocyte lineage cells. Glia 2016, 64, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, F.; Garcia, S.; Padgett, K.R.; Moraes, C.T. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012, 21, 5066–5077. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Mahad, D.J. Metabolic support of axons by oligodendrocytes: Implications for multiple sclerosis. Mult Scler Relat Disord 2014, 3, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Ziabreva, I.; Campbell, G.; Rist, J.; Zambonin, J.; Rorbach, J.; Wydro, M.M.; Lassmann, H.; Franklin, R.J.; Mahad, D. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 2010, 58, 1827–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfeld, R.; Wong, A.; Silva, J.; Li, M.; Itoh, A.; Horiuchi, M.; Itoh, T.; Pleasure, D.; Cortopassi, G. Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 2010, 10, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.T.S.; Khan, D.; Cui, Q.L.; Fuh, S.C.; Hossain, S.; Almazan, G.; Multhaup, G.; Healy, L.M.; Kennedy, T.E.; Antel, J.P. Distinct age and differentiation-state dependent metabolic profiles of oligodendrocytes under optimal and stress conditions. PLoS ONE 2017, 12, e0182372. [Google Scholar] [CrossRef] [Green Version]
- Rone, M.B.; Cui, Q.L.; Fang, J.; Wang, L.C.; Zhang, J.; Khan, D.; Bedard, M.; Almazan, G.; Ludwin, S.K.; Jones, R.; et al. Oligodendrogliopathy in Multiple Sclerosis: Low Glycolytic Metabolic Rate Promotes Oligodendrocyte Survival. J. Neurosci. 2016, 36, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Abarca, L.I.; Tabernero, A.; Medina, J.M. Oligodendrocytes use lactate as a source of energy and as a precursor of lipids. Glia 2001, 36, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.I.; Tavares, J.M.; Sonnewald, U.; Kotter, M.R. Oligodendrocytes: Development, Physiology and Glucose Metabolism. Adv. Neurobiol. 2016, 13, 275–294. [Google Scholar] [CrossRef]
- Sykes, J.E.; Lopes-Cardozo, M.; Van Den Bergh, S.G. Relationship between the pentose-phosphate pathway and the de novo synthesis of fatty acids and cholesterol in oligodendrocyte-enriched glial cultures. Neurochem. Int. 1986, 8, 77–82. [Google Scholar] [CrossRef]
- Binamé, F.; Sakry, D.; Dimou, L.; Jolivel, V.; Trotter, J. NG2 regulates directional migration of oligodendrocyte precursor cells via Rho GTPases and polarity complex proteins. J. Neurosci. 2013, 33, 10858–10874. [Google Scholar] [CrossRef] [Green Version]
- Sy, M.; Brandt, A.U.; Lee, S.U.; Newton, B.L.; Pawling, J.; Golzar, A.; Rahman, A.M.A.; Yu, Z.; Cooper, G.; Scheel, M.; et al. N-acetylglucosamine drives myelination by triggering oligodendrocyte precursor cell differentiation. J. Biol. Chem. 2020, 295, 17413–17424. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.K.; Voisin, P.; Canioni, P.; Magistretti, P.J.; Pellerin, L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb Blood Flow Metab. 2003, 23, 1298–1306. [Google Scholar] [CrossRef] [Green Version]
- Wyss, M.T.; Jolivet, R.; Buck, A.; Magistretti, P.J.; Weber, B. In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 2011, 31, 7477–7485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martín, A.; Romero-Gómez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef]
- Pellerin, L.; Bergersen, L.H.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Zhou, P.; Guan, T.; Jiang, Z.; Namaka, M.; Huang, Q.J.; Kong, J.M. Monocarboxylate transporter 1 and the vulnerability of oligodendrocyte lineage cells to metabolic stresses. CNS Neurosci. Ther. 2018, 24, 126–134. [Google Scholar] [CrossRef]
- Andres Benito, P.; Dominguez Gonzalez, M.; Ferrer, I. Altered gene transcription linked to astrocytes and oligodendrocytes in frontal cortex in Creutzfeldt-Jakob disease. Prion 2018, 12, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Li, Z.; Zhang, W.; Yao, Z. Nitric oxide might be an inducing factor in cognitive impairment in Alzheimer’s disease via downregulating the monocarboxylate transporter 1. Nitric Oxide 2019, 91, 35–41. [Google Scholar] [CrossRef]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Mironova, Y.A.; Jouroukhin, Y.; Chew, J.; Vidensky, S.; Farah, M.H.; Pletnikov, M.V.; Bergles, D.E.; Morrison, B.M.; Rothstein, J.D. MCT1 Deletion in Oligodendrocyte Lineage Cells Causes Late-Onset Hypomyelination and Axonal Degeneration. Cell Rep. 2021, 34, 108610. [Google Scholar] [CrossRef] [PubMed]
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial networks: A step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 2010, 11, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Wasseff, S.K.; Scherer, S.S. Cx32 and Cx47 mediate oligodendrocyte:astrocyte and oligodendrocyte:oligodendrocyte gap junction coupling. Neurobiol. Dis. 2011, 42, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tress, O.; Maglione, M.; May, D.; Pivneva, T.; Richter, N.; Seyfarth, J.; Binder, S.; Zlomuzica, A.; Seifert, G.; Theis, M.; et al. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J. Neurosci. 2012, 32, 7499–7518. [Google Scholar] [CrossRef]
- Hawkins, R.A.; Williamson, D.H.; Krebs, H.A. Ketone-body utilization by adult and suckling rat brain in vivo. Biochem. J. 1971, 122, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, M.A.; Krebs, H.A.; Williamson, D.H. Activities of enzymes of ketone-body utilization in brain and other tissues of suckling rats. Biochem. J. 1971, 121, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75 (Suppl. 1), 8–18. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Koper, J.W.; Lopes-Cardozo, M.; Van Golde, L.M. Preferential utilization of ketone bodies for the synthesis of myelin cholesterol in vivo. Biochim. Biophys. Acta 1981, 666, 411–417. [Google Scholar] [CrossRef]
- Edmond, J. Ketone bodies as precursors of sterols and fatty acids in the developing rat. J. Biol. Chem. 1974, 249, 72–80. [Google Scholar] [CrossRef]
- Ichihara, Y.; Doi, T.; Ryu, Y.; Nagao, M.; Sawada, Y.; Ogata, T. Oligodendrocyte Progenitor Cells Directly Utilize Lactate for Promoting Cell Cycling and Differentiation. J. Cell Physiol. 2017, 232, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Schlaphoff, L.; Depp, C.; Düking, T.; Winchenbach, J.; Neuber, J.; Ewers, D.; et al. Microglia facilitate repair of demyelinated lesions via post-squalene sterol synthesis. Nat. Neurosci. 2021, 24, 47–60. [Google Scholar] [CrossRef]
- Poduslo, S.E.; Miller, K. Ketone bodies as precursors for lipid synthesis in neurons, astrocytes, and oligodendroglia (myelin) in hyperthyroidism, hyperketonemia and hypoketonemia. Neurochem. Int. 1991, 18, 85–88. [Google Scholar] [CrossRef]
- Sykes, J.E.; Lopes-Cardozo, M.; Van Den Bergh, S.G. Substrate utilization for energy production and lipid synthesis in oligodendrocyte-enriched cultures prepared from rat brain. Neurochem. Int. 1986, 8, 67–75. [Google Scholar] [CrossRef]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Stumpf, S.K.; Berghoff, S.A.; Trevisiol, A.; Spieth, L.; Düking, T.; Schneider, L.V.; Schlaphoff, L.; Dreha-Kulaczewski, S.; Bley, A.; Burfeind, D.; et al. Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus-Merzbacher disease. Acta Neuropathol. 2019, 138, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; von Döbeln, U.; Wedell, A. The ketogenic diet compensates for AGC1 deficiency and improves myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langley, M.R.; Triplet, E.M.; Scarisbrick, I.A. Dietary influence on central nervous system myelin production, injury, and regeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165779. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Madhavarao, C.N.; Moffett, J.R.; Moore, R.A.; Viola, R.E.; Namboodiri, M.A.; Jacobowitz, D.M. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J. Comp. Neurol. 2004, 472, 318–329. [Google Scholar] [CrossRef]
- Chakraborty, G.; Mekala, P.; Yahya, D.; Wu, G.; Ledeen, R.W. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: Evidence for myelin-associated aspartoacylase. J. Neurochem. 2001, 78, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Krasnow, A.M.; Attwell, D. NMDA Receptors: Power Switches for Oligodendrocytes. Neuron 2016, 91, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Madhavarao, C.N.; Arun, P.; Moffett, J.R.; Szucs, S.; Surendran, S.; Matalon, R.; Garbern, J.; Hristova, D.; Johnson, A.; Jiang, W.; et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan;s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 5221–5226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Leone, P.; Wu, G.; Francis, J.S.; Li, H.; Jain, M.R.; Serikawa, T.; Ledeen, R.W. Myelin lipid abnormalities in the aspartoacylase-deficient tremor rat. Neurochem. Res. 2009, 34, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Amaral, I.A.; Hadera, M.G.; Kotter, M.; Sonnewald, U. Oligodendrocytes Do Not Export NAA-Derived Aspartate In Vitro. Neurochem. Res. 2017, 42, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, J.S.; Strande, L.; Markov, V.; Leone, P. Aspartoacylase supports oxidative energy metabolism during myelination. J. Cereb Blood Flow Metab. 2012, 32, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Von Jonquieres, G.; Spencer, Z.H.T.; Rowlands, B.D.; Klugmann, C.B.; Bongers, A.; Harasta, A.E.; Parley, K.E.; Cederholm, J.; Teahan, O.; Pickford, R.; et al. Uncoupling N-acetylaspartate from brain pathology: Implications for Canavan disease gene therapy. Acta Neuropathol. 2018, 135, 95–113. [Google Scholar] [CrossRef] [Green Version]
- Shields, S.A.; Gilson, J.M.; Blakemore, W.F.; Franklin, R.J. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 1999, 28, 77–83. [Google Scholar] [CrossRef]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [CrossRef]
- Mei, F.; Lehmann-Horn, K.; Shen, Y.A.; Rankin, K.A.; Stebbins, K.J.; Lorrain, D.S.; Pekarek, K.; A Sagan, S.; Xiao, L.; Teuscher, C.; et al. Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. Elife 2016, 5, e18246. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef]
- Cui, Q.L.; Khan, D.; Rone, M.; Rao, T.S.V.; Johnson, R.M.; Lin, Y.H.; Bilodeau, P.A.; Hall, J.A.; Rodriguez, M.; Kennedy, T.E.; et al. Sublethal oligodendrocyte injury: A reversible condition in multiple sclerosis? Ann. Neurol. 2017, 81, 811–824. [Google Scholar] [CrossRef]
- Duncan, I.D.; Radcliff, A.B.; Heidari, M.; Kidd, G.; August, B.K.; Wierenga, L.A. The adult oligodendrocyte can participate in remyelination. Proc. Natl. Acad. Sci. USA 2018, 115, E11807–E11816. [Google Scholar] [CrossRef] [Green Version]
- Heß, K.; Starost, L.; Kieran, N.W.; Thomas, C.; Vincenten, M.C.J.; Antel, J.; Martino, G.; Huitinga, I.; Healy, L.; Kuhlmann, T. Lesion stage-dependent causes for impaired remyelination in MS. Acta Neuropathol. 2020, 140, 359–375. [Google Scholar] [CrossRef]
- Schiepers, C.; Van Hecke, P.; Vandenberghe, R.; Van Oostende, S.; Dupont, P.; Demaerel, P.; Bormans, G.; Carton, H. Positron emission tomography, magnetic resonance imaging and proton NMR spectroscopy of white matter in multiple sclerosis. Mult. Scler. 1997, 3, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.G.F.; Luo, J.X.X.; Cui, Q.L.; Perlman, K.; Pernin, F.; Yaqubi, M.; Hall, J.A.; Dudley, R.; Srour, M.; Couturier, C.P.; et al. Age-related injury responses of human oligodendrocytes to metabolic insults: Link to BCL-2 and autophagy pathways. Commun. Biol. 2021, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwin, S.K.; Johnson, E.S. Evidence for a “dying-back” gliopathy in demyelinating disease. Ann. Neurol. 1981, 9, 301–305. [Google Scholar] [CrossRef]
- Duncan, I.D.; Brower, A.; Kondo, Y.; Curlee, J.F.; Schultz, R.D. Extensive remyelination of the CNS leads to functional recovery. Proc. Natl. Acad. Sci. USA 2009, 106, 6832–6836. [Google Scholar] [CrossRef] [Green Version]
- Klosinski, L.P.; Yao, J.; Yin, F.; Fonteh, A.N.; Harrington, M.G.; Christensen, T.A.; Trushina, E.; Brinton, R.D. White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer’s Disease. EBioMedicine 2015, 2, 1888–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tepavčević, V. Oligodendroglial Energy Metabolism and (re)Myelination. Life 2021, 11, 238. https://doi.org/10.3390/life11030238
Tepavčević V. Oligodendroglial Energy Metabolism and (re)Myelination. Life. 2021; 11(3):238. https://doi.org/10.3390/life11030238
Chicago/Turabian StyleTepavčević, Vanja. 2021. "Oligodendroglial Energy Metabolism and (re)Myelination" Life 11, no. 3: 238. https://doi.org/10.3390/life11030238
APA StyleTepavčević, V. (2021). Oligodendroglial Energy Metabolism and (re)Myelination. Life, 11(3), 238. https://doi.org/10.3390/life11030238