
Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Insulin Receptor in the Brain
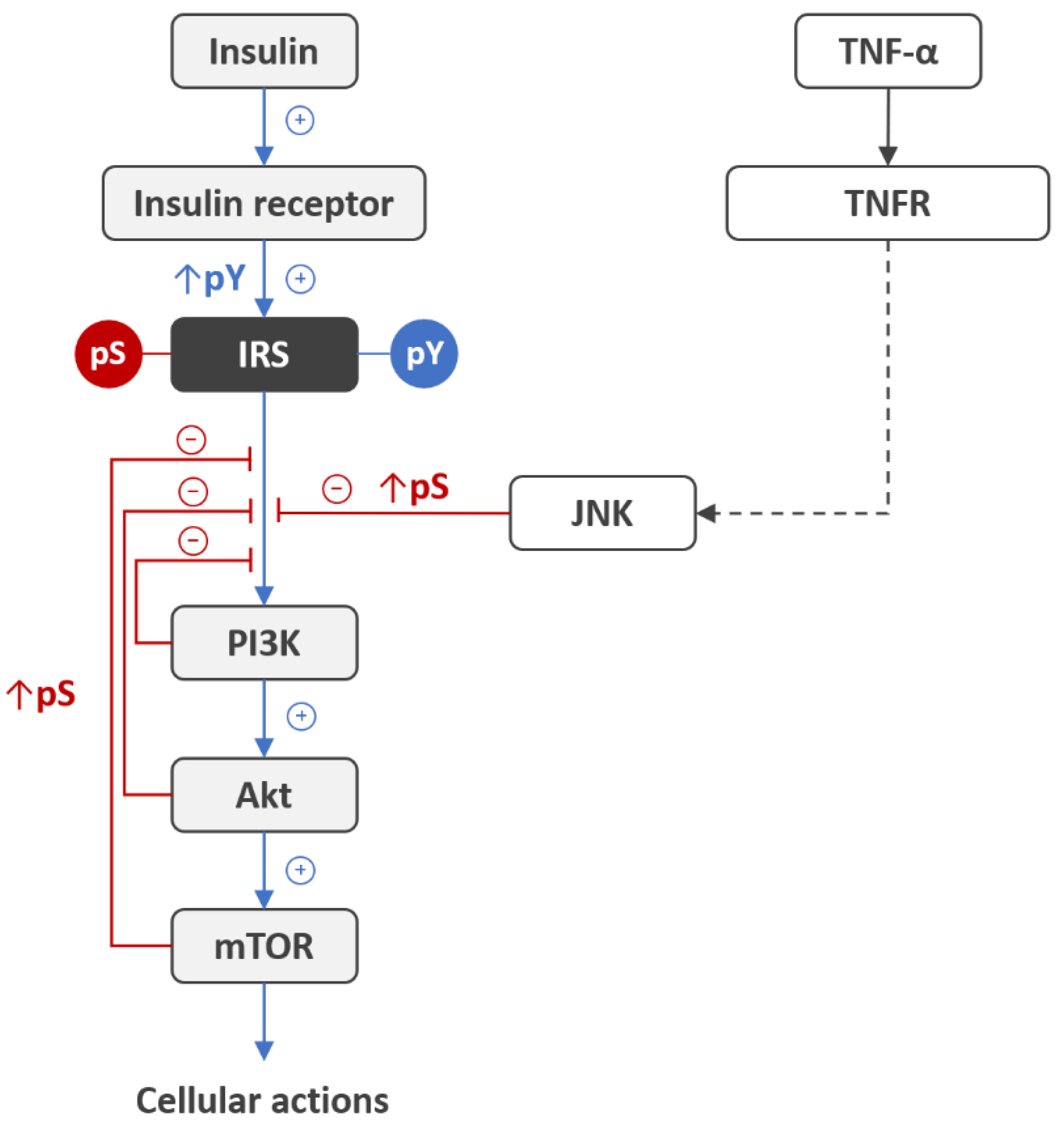
3. Insulin Receptor Activation: A Role of Redox Priming
4. Critical Role of Redox Signaling in the Activation of the Neuronal Insulin Receptor
5. G Protein Activity in the Activation of the Neuronal Insulin Receptor
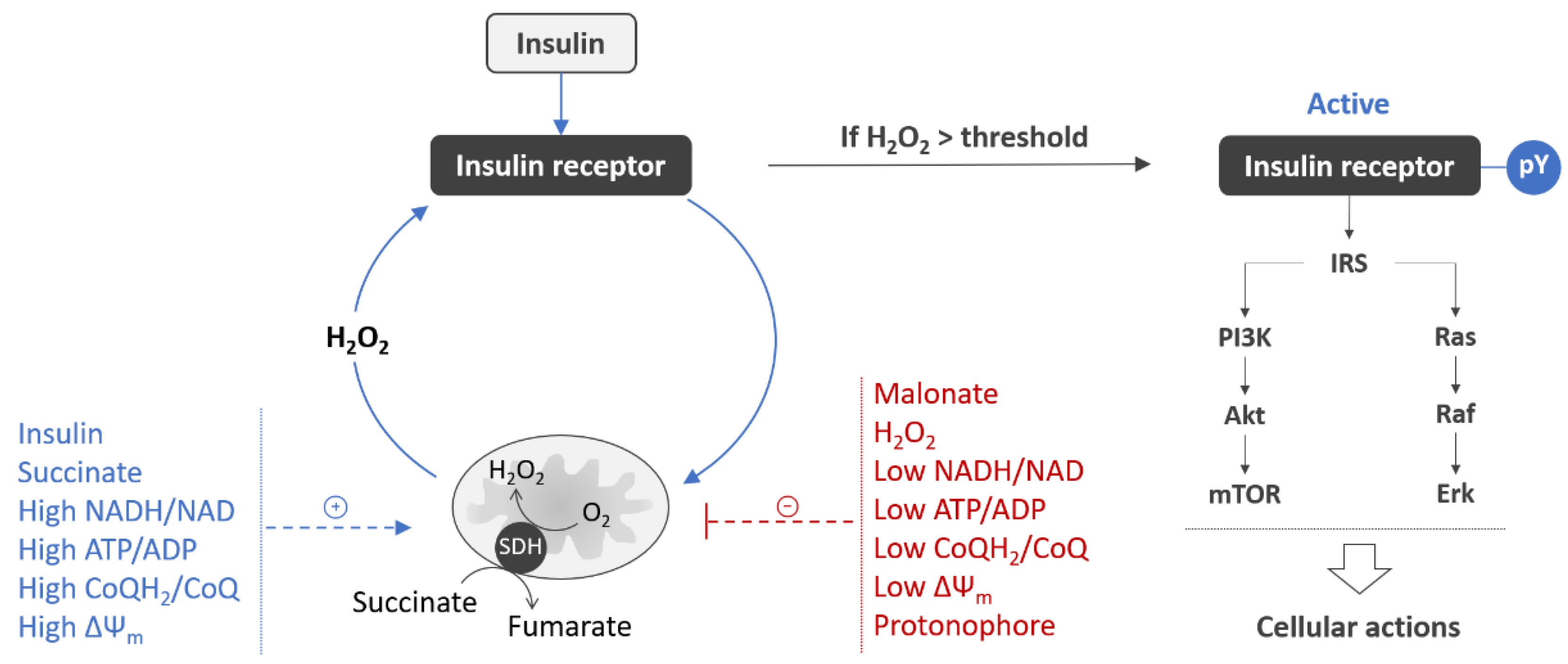
6. Mitochondrial Signaling Is an Integral Part of the Insulin Receptor Activation Process in Neurons
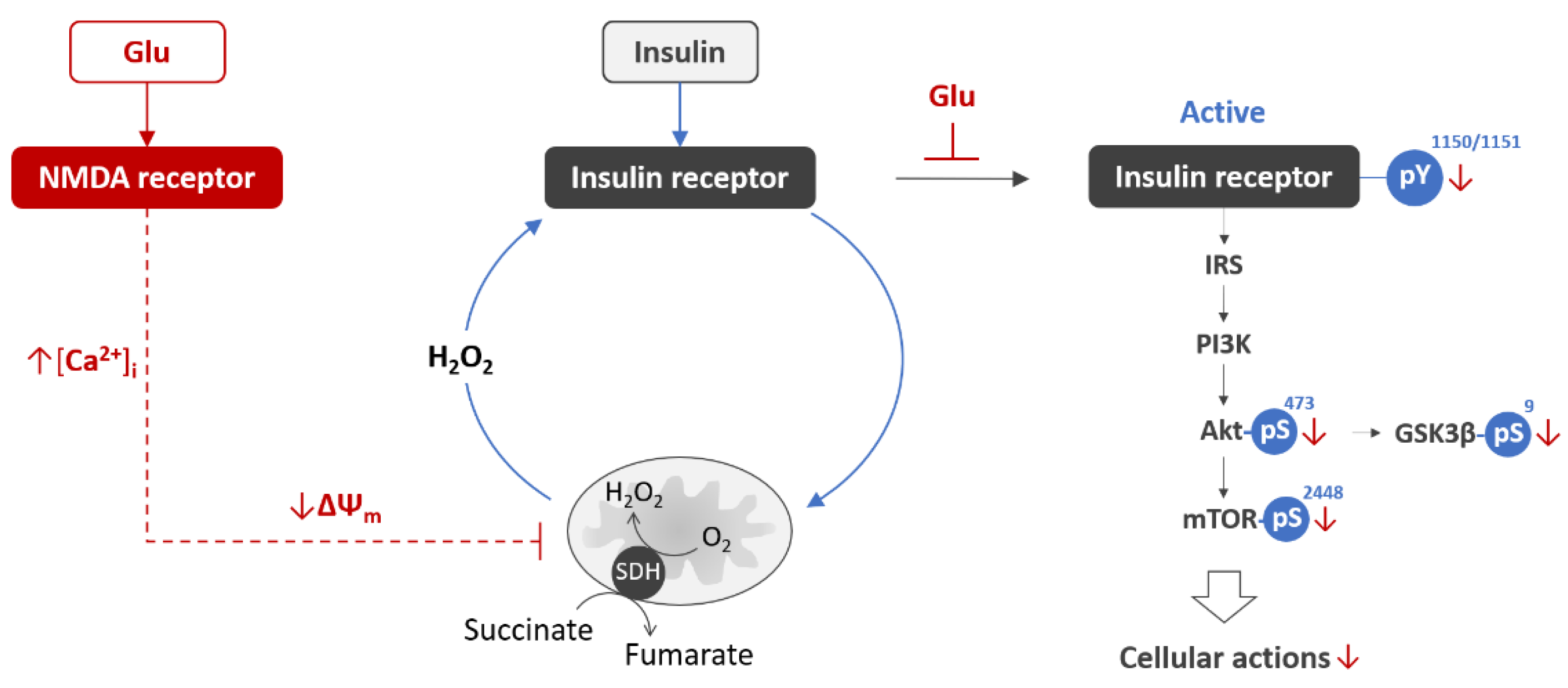
7. Glutamate Excitotoxicity Impairs Activation of the Neuronal Insulin Receptor
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Himsworth, H.P. Diabetes mellitus: Its differentiation into insulin-sensitive and insulin-insensitive types. Int. J. Epidemiol. 2013, 42, 1594–1598. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. Consensus Development Conference on Insulin Resistance. 5–6 November 1997. Diabetes Care 1998, 21, 310–314. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog. Neurobiol. 2016, 145–146, 98–120. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Sripetchwandee, J.; Chattipakorn, N.; Chattipakorn, S.C. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front. Endocrinol. 2018, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Lyra e Silva, N.D.M.; Lam, M.P.; Soares, C.N.; Munoz, D.P.; Milev, R.; De Felice, F.G. Insulin resistance as a shared pathogenic mechanism between depression and type 2 diabetes. Front. Psychiatry 2019, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Hamer, J.; Testani, D.; Mansur, R.; Lee, Y.; Subramaniapillai, M.; McIntyre, R. Brain insulin resistance: A treatment target for cognitive impairment and anhedonia in depression. Exp. Neurol. 2019, 315, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Karelina, K.; Sarac, B.; Freeman, L.M.; Gaier, K.R.; Weil, Z.M. Traumatic brain injury and obesity induce persistent central insulin resistance. Eur. J. Neurosci. 2016, 43, 1034–1043. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Role of the blood-brain barrier in central nervous system insulin resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [Green Version]
- Frazier, H.N.; Ghoweri, A.O.; Anderson, K.L.; Lin, R.L.; Porter, N.M.; Thibault, O. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol. 2019, 313, 79–87. [Google Scholar] [CrossRef]
- Rajasekar, N.; Nath, C.; Hanif, K.; Shukla, R. Intranasal insulin administration ameliorates streptozotocin (ICV)-induced insulin receptor dysfunction, neuroinflammation, amyloidogenesis, and memory impairment in rats. Mol. Neurobiol. 2017, 54, 6507–6522. [Google Scholar] [CrossRef] [PubMed]
- Freiherr, J.; Hallschmid, M.; Frey, W.H., 2nd; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilden, P.A.; Kahn, C.R.; Siddle, K.; White, M.F. Insulin receptor kinase domain autophosphorylation regulates receptor enzymatic function. J. Biol. Chem. 1992, 267, 16660–16668. [Google Scholar] [CrossRef]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E581–E591. [Google Scholar] [CrossRef] [Green Version]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [Green Version]
- Hançer, N.J.; Qiu, W.; Cherella, C.; Li, Y.; Copps, K.D.; White, M.F. Insulin and metabolic stress stimulate multisite serine/threonine phosphorylation of insulin receptor substrate 1 and inhibit tyrosine phosphorylation. J. Biol. Chem. 2014, 289, 12467–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Toledo, J.B.; Lee, E.B.; Arvanitakis, Z.; Kazi, H.; Han, L.Y.; Louneva, N.; Lee, V.M.; Kim, S.F.; Trojanowski, J.Q.; et al. Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol. 2014, 128, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Ono, H. Molecular Mechanisms of Hypothalamic Insulin Resistance. Int. J. Mol. Sci. 2019, 20, 1317. [Google Scholar] [CrossRef] [Green Version]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, H.K.; Zierath, J.R. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem. Biophys. 2007, 48, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [PubMed] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Cardoso, S.M.; Correia, S.C.; Moreira, P.I. Tortuous Paths of Insulin Signaling and Mitochondria in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1128, 161–183. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Unger, J.; McNeill, T.H.; Moxley, R.T., 3rd; White, M.; Moss, A.; Livingston, J.N. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 1989, 31, 143–157. [Google Scholar] [CrossRef]
- Abbott, M.A.; Wells, D.G.; Fallon, J.R. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J. Neurosci. 1999, 19, 7300–7308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, K.A.; Zahniser, N.R.; Berhanu, P.; Brandenburg, D.; Olefsky, J.M. Structural differences between insulin receptors in the brain and peripheral target tissues. J. Biol. Chem. 1983, 258, 8527–8530. [Google Scholar] [CrossRef]
- Gammeltoft, S.; Fehlmann, M.; Van Obberghen, E. Insulin receptors in the mammalian central nervous system: Binding characteristics and subunit structure. Biochimie 1985, 67, 1147–1153. [Google Scholar] [CrossRef]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain 2015, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, Y.; Flier, J.S.; Benecke, H.; Ransil, B.J.; Moller, D.E. Ligand-binding properties of the two isoforms of the human insulin receptor. Endocrinology 1993, 132, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar] [CrossRef] [Green Version]
- Mosthaf, L.; Grako, K.; Dull, T.J.; Coussens, L.; Ullrich, A.; McClain, D.A. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. 1990, 9, 2409–2413. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Flier, J.S.; Yokota, A.; Benecke, H.; Backer, J.M.; Moller, D.E. Functional properties of two naturally occurring isoforms of the human insulin receptor in Chinese hamster ovary cells. Endocrinology 1991, 129, 2058–2066. [Google Scholar] [CrossRef]
- Ziegler, A.N.; Schneider, J.S.; Qin, M.; Tyler, W.A.; Pintar, J.E.; Fraidenraich, D.; Wood, T.L.; Levison, S.W. IGF-II promotes stemness of neural restricted precursors. Stem Cells 2012, 30, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.N.; Chidambaram, S.; Forbes, B.E.; Wood, T.L.; Levison, S.W. Insulin-like growth factor-II (IGF-II) and IGF-II analogs with enhanced insulin receptor-a binding affinity promote neural stem cell expansion. J. Biol. Chem. 2014, 289, 4626–4633. [Google Scholar] [CrossRef] [Green Version]
- van der Heide, L.P.; Ramakers, G.M.; Smidt, M.P. Insulin signaling in the central nervous system: Learning to survive. Prog. Neurobiol. 2006, 79, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.J.; Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Insulin, PKC signaling pathways and synaptic remodeling during memory storage and neuronal repair. Eur. J. Pharm. 2008, 585, 76–87. [Google Scholar] [CrossRef]
- Ferrario, C.R.; Reagan, L.P. Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology 2018, 136, 182–191. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Brown, J.C., 3rd; Webster, W.W.; Morrisett, R.A.; Monaghan, D.T. Insulin potentiates N-methyl-D-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neurosci. Lett. 1995, 192, 5–8. [Google Scholar] [CrossRef]
- Chen, C.; Leonard, J.P. Protein tyrosine kinase-mediated potentiation of currents from cloned NMDA receptors. J. Neurochem. 1996, 67, 194–200. [Google Scholar] [CrossRef]
- Christie, J.M.; Wenthold, R.J.; Monaghan, D.T. Insulin causes a transient tyrosine phosphorylation of NR2A and NR2B NMDA receptor subunits in rat hippocampus. J. Neurochem. 1999, 72, 1523–1528. [Google Scholar] [CrossRef]
- Liao, G.Y.; Leonard, J.P. Insulin modulation of cloned mouse NMDA receptor currents in Xenopus oocytes. J. Neurochem. 1999, 73, 1510–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.L.; Leonard, J.P. PKC site mutations reveal differential modulation by insulin of NMDA receptors containing NR2A or NR2B subunits. J. Neurochem. 2005, 92, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Xiong, Z.G.; Man, H.Y.; Ackerley, C.A.; Braunton, J.; Lu, W.Y.; Becker, L.E.; MacDonald, J.F.; Wang, Y.T. Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 1997, 388, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Jin, Y.; Kumar-Mendu, S.; Degerman, E.; Groop, L.; Birnir, B. Insulin reduces neuronal excitability by turning on GABA(A) channels that generate tonic current. PLoS ONE 2011, 6, e16188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNay, E.C.; Recknagel, A.K. Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol. Learn. Mem. 2011, 96, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 1999, 57, 693–705. [Google Scholar] [CrossRef]
- Komori, T.; Morikawa, Y.; Tamura, S.; Doi, A.; Nanjo, K.; Senba, E. Subcellular localization of glucose transporter 4 in the hypothalamic arcuate nucleus of ob/ob mice under basal conditions. Brain Res. 2005, 1049, 34–42. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Hendry, R.M.; Reagan, L.P. Insulin-stimulated translocation of GLUT4 to the plasma membrane in rat hippocampus is PI3-kinase dependent. Brain Res. 2009, 1296, 35–45. [Google Scholar] [CrossRef] [Green Version]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Pearson-Leary, J.; Jahagirdar, V.; Sage, J.; McNay, E.C. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 2018, 338, 32–39. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Wei, L.; Hubbard, S.R.; Hendrickson, W.A.; Ellis, L. Expression, characterization, and crystallization of the catalytic core of the human insulin receptor protein-tyrosine kinase domain. J. Biol. Chem. 1995, 270, 8122–8130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, L.; Clauser, E.; Morgan, D.O.; Edery, M.; Roth, R.A.; Rutter, W.J. Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell 1986, 45, 721–732. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar] [CrossRef]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997, 16, 5572–5581. [Google Scholar] [CrossRef]
- Schmid, E.; El Benna, J.; Galter, D.; Klein, G.; Dröge, W. Redox priming of the insulin receptor beta-chain associated with altered tyrosine kinase activity and insulin responsiveness in the absence of tyrosine autophosphorylation. FASEB J. 1998, 12, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Schmid, E.; Hotz-Wagenblatt, A.; Hacj, V.; Dröge, W. Phosphorylation of the insulin receptor kinase by phosphocreatine in combination with hydrogen peroxide: The structural basis of redox priming. FASEB J. 1999, 13, 1491–1500. [Google Scholar] [CrossRef]
- May, J.M.; de Haën, C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J. Biol. Chem. 1979, 254, 2214–2220. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Lane, R.H.; Lynn, W.S. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem. Pharm. 1978, 27, 2589–2594. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Mahadev, K.; Wu, X. Redox paradox: Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 2005, 54, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Flohé, L.; Loschen, G.; Günzler, W.A.; Eichele, E. Glutathione peroxidase, V. The kinetic mechanism. Hoppe Seylers Z. Physiol. Chem. 1972, 353, 987–999. [Google Scholar] [CrossRef]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, D.T.; Stults, F.H.; Tappel, A.L. Purification and properties of rat lung soluble glutathione peroxidase. Biochim. Biophys. Acta 1976, 445, 558–566. [Google Scholar] [CrossRef]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Krieger-Brauer, H.I.; Medda, P.K.; Kather, H. Insulin-induced activation of NADPH-dependent H2O2 generation in human adipocyte plasma membranes is mediated by Galphai2. J. Biol. Chem. 1997, 272, 10135–10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadev, K.; Wu, X.; Zilbering, A.; Zhu, L.; Lawrence, J.T.; Goldstein, B.J. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J. Biol. Chem. 2001, 276, 48662–48669. [Google Scholar] [CrossRef] [Green Version]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomytkin, I.A.; Kolesova, O.E. Effect of insulin on the rate of hydrogen peroxide generation in mitochondria. Bull. Exp. Biol. Med. 2003, 135, 541–542. [Google Scholar] [CrossRef]
- Pomytkin, I.A.; Kolesova, O.E. Key role of succinate dehydrogenase in insulin-induced inactivation of protein tyrosine phosphatases. Bull. Exp. Biol. Med. 2002, 133, 568–570. [Google Scholar] [CrossRef]
- Jaillard, T.; Roger, M.; Galinier, A.; Guillou, P.; Benani, A.; Leloup, C.; Casteilla, L.; Pénicaud, L.; Lorsignol, A. Hypothalamic reactive oxygen species are required for insulin-induced food intake inhibition: An NADPH oxidase-dependent mechanism. Diabetes 2009, 58, 1544–1549. [Google Scholar] [CrossRef] [Green Version]
- Drougard, A.; Fournel, A.; Valet, P.; Knauf, C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storozhevykh, T.P.; Senilova, Y.E.; Persiyantseva, N.A.; Pinelis, V.G.; Pomytkin, I.A. Mitochondrial respiratory chain is involved in insulin-stimulated hydrogen peroxide production and plays an integral role in insulin receptor autophosphorylation in neurons. BMC Neurosci. 2007, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persiyantseva, N.A.; Storozhevykh, T.P.; Senilova, Y.E.; Gorbacheva, L.R.; Pinelis, V.G.; Pomytkin, I.A. Mitochondrial H2O2 as an enable signal for triggering autophosphorylation of insulin receptor in neurons. J. Mol. Signal. 2013, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Raina, A.K.; Lee, H.G.; Casadesus, G.; Smith, M.A.; Perry, G. Oxidative stress signalling in Alzheimer’s disease. Brain Res. 2004, 1000, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Zhu, X.; Liu, Q.; Honda, K.; Siedlak, S.L.; Harris, P.L.; Smith, M.A.; Perry, G. Compensatory responses induced by oxidative stress in Alzheimer disease. Biol. Res. 2006, 39, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aksenov, M.Y.; Tucker, H.M.; Nair, P.; Aksenova, M.V.; Butterfield, D.A.; Estus, S.; Markesbery, W.R. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer’s disease. J. Mol. Neurosci. 1998, 11, 151–164. [Google Scholar] [CrossRef]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J. Neural. Transm. Suppl. 2001, 61, 223–235. [Google Scholar] [CrossRef]
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef]
- Sultana, R.; Boyd-Kimball, D.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Butterfield, D.A. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J. Alzheimers Dis. 2007, 11, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeliga, M. Peroxiredoxins in Neurodegenerative Diseases. Antioxidants 2020, 9, 1203. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, J.; Nürnberg, B.; Krieger-Brauer, H.I. Ligand-dependent autophosphorylation of the insulin receptor is positively regulated by Gi-proteins. Biochem. J. 2004, 380 Pt 3, 831–836. [Google Scholar] [CrossRef]
- Song, X.; Zheng, X.; Malbon, C.C.; Wang, H. Galpha i2 enhances in vivo activation of and insulin signaling to GLUT4. J. Biol. Chem. 2001, 276, 34651–34658. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.F.; Guo, J.H.; Moxham, C.M.; Wang, H.Y.; Malbon, C.C. Conditional, tissue-specific expression of Q205L G alpha i2 in vivo mimics insulin action. J. Mol. Med. 1997, 75, 283–289. [Google Scholar] [CrossRef]
- Tao, J.; Malbon, C.C.; Wang, H.Y. Galpha(i2) enhances insulin signaling via suppression of protein-tyrosine phosphatase 1B. J. Biol. Chem. 2001, 276, 39705–39712. [Google Scholar] [CrossRef] [Green Version]
- Moxham, C.M.; Malbon, C.C. Insulin action impaired by deficiency of the G-protein subunit G ialpha2. Nature 1996, 379, 840–844. [Google Scholar] [CrossRef]
- Kato, K.; Kurobe, N.; Suzuki, F.; Morishita, R.; Asano, T.; Sato, T.; Inagaki, T. Concentrations of several proteins characteristic of nervous tissue in cerebral cortex of patients with Alzheimer’s disease. J. Mol. Neurosci. 1991, 3, 95–99. [Google Scholar] [CrossRef]
- McLaughlin, M.; Ross, B.M.; Milligan, G.; McCulloch, J.; Knowler, J.T. Robustness of G proteins in Alzheimer’s disease: An immunoblot study. J. Neurochem. 1991, 57, 9–14. [Google Scholar] [CrossRef]
- Storozheva, Z.I.; Proshin, A.T.; Sherstnev, V.V.; Storozhevykh, T.P.; Senilova, Y.E.; Persiyantseva, N.A.; Pinelis, V.G.; Semenova, N.A.; Zakharova, E.I.; Pomytkin, I.A. Dicholine salt of succinic acid, a neuronal insulin sensitizer, ameliorates cognitive deficits in rodent models of normal aging, chronic cerebral hypoperfusion, and beta-amyloid peptide-(25–35)-induced amnesia. BMC Pharm. 2008, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cino, M.; Del Maestro, R.F. Generation of hydrogen peroxide by brain mitochondria: The effect of reoxygenation following postdecapitative ischemia. Arch. Biochem. Biophys. 1989, 269, 623–638. [Google Scholar] [CrossRef]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Bessman, S.P.; Mohan, C. Insulin as a probe of mitochondrial metabolism in situ. Mol. Cell Biochem. 1997, 174, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Bessman, S.P.; Mohan, C.; Zaidise, I. Intracellular site of insulin action: Mitochondrial Krebs cycle. Proc. Natl. Acad. Sci. USA 1986, 83, 5067–5070. [Google Scholar] [CrossRef] [Green Version]
- Ackrell, B.A.; Kearney, E.B.; Singer, T.P. Mammalian succinate dehydrogenase. Methods Enzymol. 1978, 53, 466–483. [Google Scholar] [CrossRef]
- Gutman, M.; Kearney, E.B.; Singer, T.P. Control of succinate dehydrogenase in mitochondria. Biochemistry 1971, 10, 4763–4770. [Google Scholar] [CrossRef] [PubMed]
- Gutman, M.; Kearney, E.B.; Singer, T.P. Activation of succinate dehydrogenase by electron flux from NADH and its possible regulatory function. Biochem. Biophys. Res. Commun. 1971, 42, 1016–1023. [Google Scholar] [CrossRef]
- Gutman, M.; Kearney, E.B.; Singer, T.P. Multiple control mechanisms for succinate dehydrogenase in mitochondria. Biochem. Biophys. Res. Commun. 1971, 44, 526–532. [Google Scholar] [CrossRef]
- Nulton-Persson, A.C.; Szweda, L.I. Modulation of mitochondrial function by hydrogen peroxide. J. Biol. Chem 2001, 276, 23357–23361. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.D.; Matsuzaki, S.; Humphries, K.M. Inhibition of succinate-linked respiration and complex II activity by hydrogen peroxide. Arch. Biochem. Biophys. 2009, 488, 69–75. [Google Scholar] [CrossRef]
- Gutman, M. Modulation of mitochondrial succinate dehydrogenase activity, mechanism and function. Mol. Cell Biochem. 1978, 20, 41–60. [Google Scholar] [CrossRef]
- Ackrell, B.A.; Kearney, E.B.; Edmondson, D. Mechanism of the reductive activation of succinate dehydrogenase. J. Biol. Chem. 1975, 250, 7114–7119. [Google Scholar] [CrossRef]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.; Mina, E.; Glabe, C.; Busciglio, J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 2006, 26, 6011–6018. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharm. 2012, 167, 699–719. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Okamoto, K.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Willard, S.S.; Koochekpour, S. Glutamate, glutamate receptors, and downstream signaling pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Khodorov, B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog. Biophys. Mol. Biol. 2004, 86, 279–351. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tymianski, M.; Charlton, M.P.; Carlen, P.L.; Tator, C.H. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993, 607, 319–323. [Google Scholar] [CrossRef]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 2012, 46, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal glutamate excitotoxicity. Biochim. Biophys. Acta 1998, 1366, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Krasil’nikova, I.; Surin, A.; Sorokina, E.; Fisenko, A.; Boyarkin, D.; Balyasin, M.; Demchenko, A.; Pomytkin, I.; Pinelis, V. Insulin Protects Cortical Neurons Against Glutamate Excitotoxicity. Front. Neurosci. 2019, 13, 1027. [Google Scholar] [CrossRef] [Green Version]
- Cui, A.L.; Zhang, Y.H.; Li, J.Z.; Song, T.; Liu, X.M.; Wang, H.; Zhang, C.; Ma, G.L.; Zhang, H.; Li, K. Humanin rescues cultured rat cortical neurons from NMDA-induced toxicity through the alleviation of mitochondrial dysfunction. Drug Des. Devel. 2017, 11, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr. Mol. Med. 2004, 4, 149–177. [Google Scholar] [CrossRef]
- Pomytkin, I.; Krasil’nikova, I.; Bakaeva, Z.; Surin, A.; Pinelis, V. Excitotoxic glutamate causes neuronal insulin resistance by inhibiting insulin receptor/Akt/mTOR pathway. Mol. Brain 2019, 12, 112. [Google Scholar] [CrossRef]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional synapse in Alzheimer’s disease—A focus on NMDA receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharm. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a Target Against Neurodegenerative Processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Olloquequi, J.; Cornejo-Córdova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of glutamate toxicity in multiple sclerosis: Biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Datusalia, A.K.; Agarwal, P.; Singh, J.N.; Sharma, S.S. Hyper-insulinemia increases the glutamate-excitotoxicity in cortical neurons: A mechanistic study. Eur. J. Pharm. 2018, 833, 524–530. [Google Scholar] [CrossRef]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [Green Version]
- Thibault, O.; Anderson, K.L.; DeMoll, C.; Brewer, L.D.; Landfield, P.W.; Porter, N.M. Hippocampal calcium dysregulation at the nexus of diabetes and brain aging. Eur. J. Pharm. 2013, 719, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; van der Heide, L.P.; Kamal, A.; Bleys, R.L.; Gispen, W.H. Ageing and diabetes: Implications for brain function. Eur. J. Pharm. 2002, 441, 1–14. [Google Scholar] [CrossRef]
- Maimaiti, S.; Frazier, H.N.; Anderson, K.L.; Ghoweri, A.O.; Brewer, L.D.; Porter, N.M.; Thibault, O. Novel calcium-related targets of insulin in hippocampal neurons. Neuroscience 2017, 364, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pomytkin, I.; Pinelis, V. Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life 2021, 11, 262. https://doi.org/10.3390/life11030262
Pomytkin I, Pinelis V. Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life. 2021; 11(3):262. https://doi.org/10.3390/life11030262
Chicago/Turabian StylePomytkin, Igor, and Vsevolod Pinelis. 2021. "Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions" Life 11, no. 3: 262. https://doi.org/10.3390/life11030262
APA StylePomytkin, I., & Pinelis, V. (2021). Brain Insulin Resistance: Focus on Insulin Receptor-Mitochondria Interactions. Life, 11(3), 262. https://doi.org/10.3390/life11030262