
Two Decades of Evolution of Our Understanding of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Characterization of TRPM2 in a Cell-Free System and Identification of Direct Effectors
2.1. Activation of TRPM2 by ADPR, Ca2+ and Phosphatidylinositol-4,5-bisphosphate (PIP2)
2.2. Pyridine Nucleotides and Their Derivatives Are Not Direct Activators of TRPM2
3. Gating Properties of the TRPM2 Channel
3.1. hsTRPM2 Is Not an Active ADPR Hydrolase
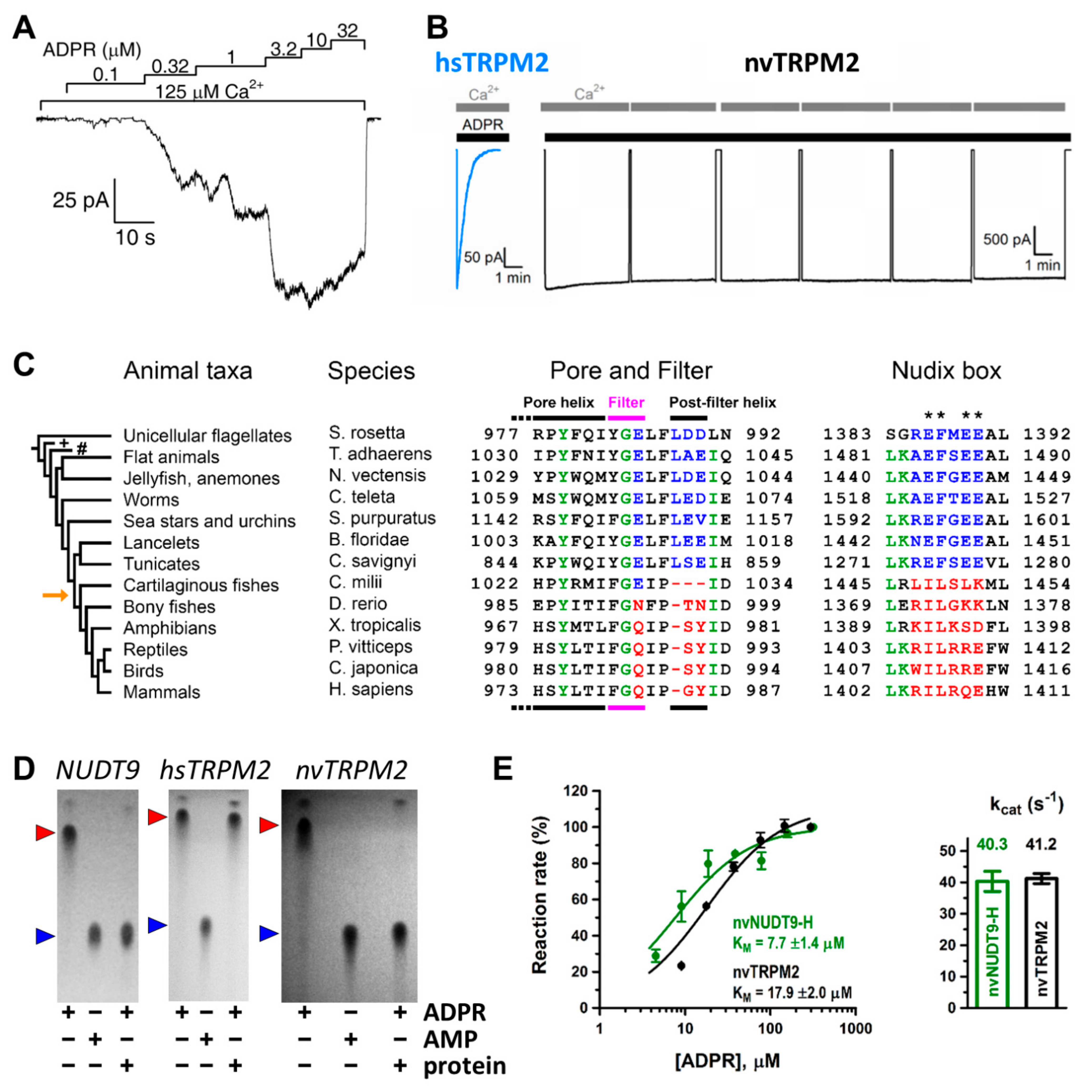
3.2. Invertebrate TRPM2 Channels Are Active ADPRase Chanzymes
4. Structure of TRPM2 Solved by Single Particle Cryo-EM
4.1. The Resolution Revolution of Single-Particle Cryo-EM
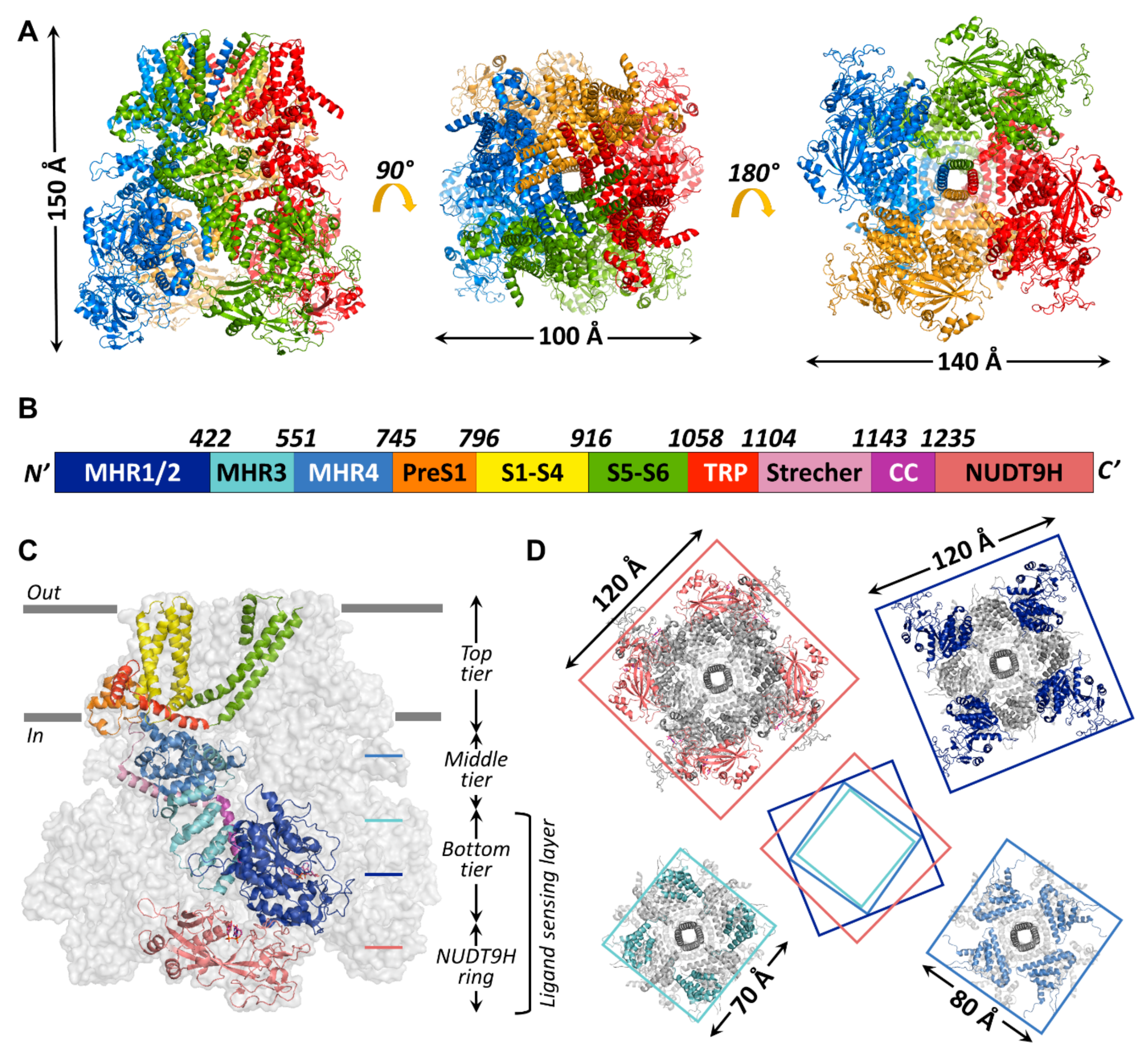
4.2. General Assembly of the TRPM2 Channel
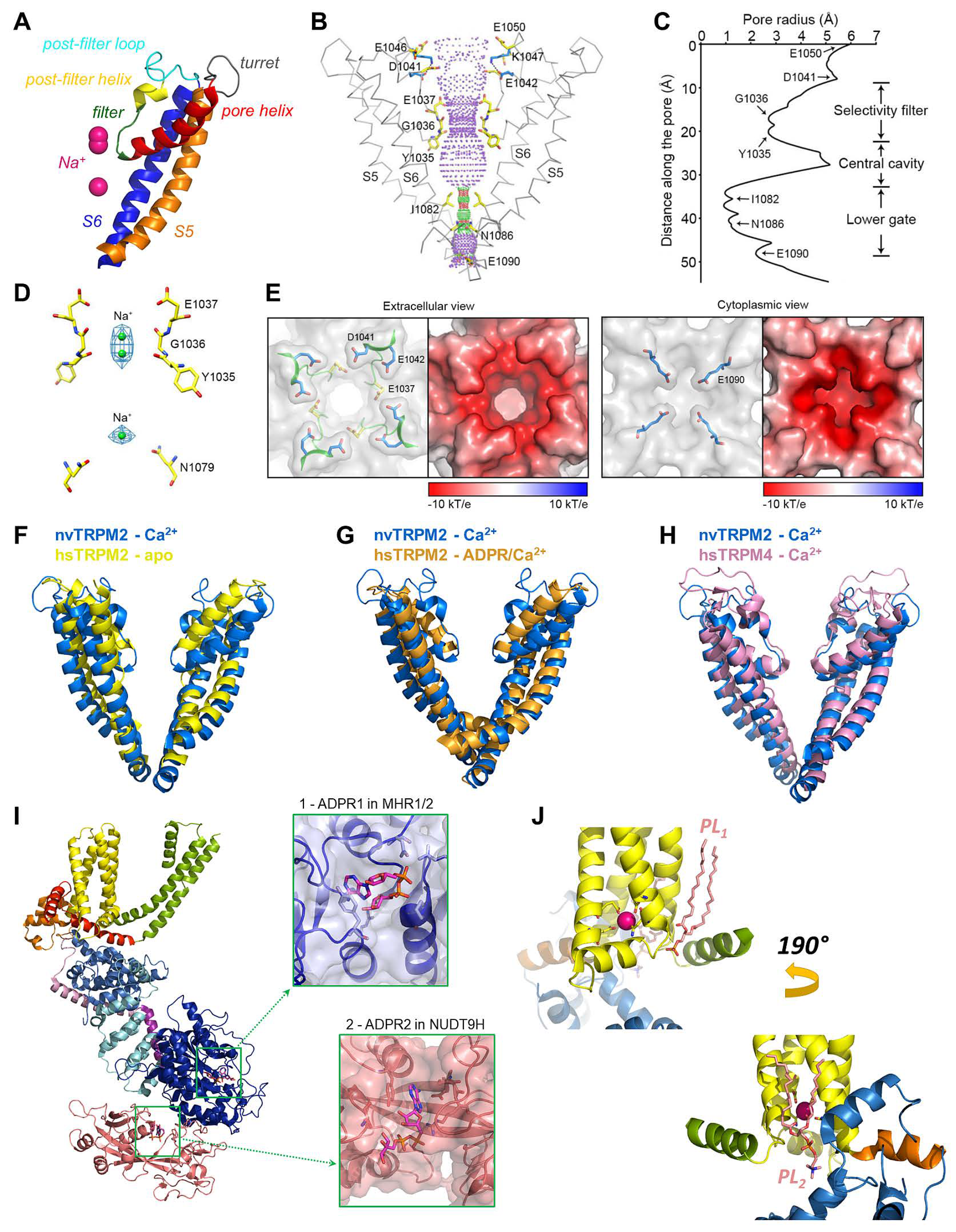
4.3. The TMD Region and Pore of the TRPM2 Channel

4.4. The Ca2+ and PIP2 Binding Sites of the TRPM2 Channel
4.5. The ADPR Binding Sites and Molecular Gating of the TRPM2 Channel
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Siemens, J. TRP ion channels in thermosensation, thermoregulation and metabolism. Temperature 2015, 2, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.-L.; Scharenberg, A.M.; Penner, R.; Kinet, J.-P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Liman, E.R. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. USA 2003, 100, 15160–15165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Kühn, F.J. Structure-Function Relationship of TRPM2: Recent Advances, Contradictions, and Open Questions. Int. J. Mol. Sci. 2020, 21, 6481. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nat. Cell Biol. 2001, 411, 595–599. [Google Scholar] [CrossRef]
- Uchida, K.; Tominaga, M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion. Endocr. J. 2011, 58, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflam-matory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef]
- Song, K.; Wang, H.; Kamm, G.B.; Pohle, J.; Reis, F.D.C.; Heppenstall, P.; Wende, H.; Siemens, J. The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 2016, 353, 1393–1398. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G.; Voets, T.; Peters, J.A. Transient Receptor Potential Cation Channels in Disease. Physiol. Rev. 2007, 87, 165–217. [Google Scholar] [CrossRef] [Green Version]
- Fonfria, E.; Marshall, I.C.B.; Boyfield, I.; Skaper, S.D.; Hughes, J.P.; E Owen, D.; Zhang, W.; Miller, B.A.; Benham, C.D.; McNulty, E.S. Amyloid beta-peptide (1–42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 2005, 95, 715–723. [Google Scholar] [CrossRef]
- Hermosura, M.C.; Cui, A.M.; Go, R.C.V.; Davenport, B.; Shetler, C.M.; Heizer, J.W.; Schmitz, C.; Mocz, G.; Garruto, R.M.; Perraud, A.-L. Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc. Natl. Acad. Sci. USA 2008, 105, 18029–18034. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, S.; Kawakami, S.; Hara, Y.; Wakamori, M.; Itoh, E.; Minami, T.; Takada, Y.; Kume, T.; Katsuki, H.; Mori, Y.; et al. A Critical Role of TRPM2 in Neuronal Cell Death by Hydrogen Peroxide. J. Pharmacol. Sci. 2006, 101, 66–76. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Crystal Structure of a Mammalian Voltage-Dependent Shaker Family K+ Channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Voltage Sensor of Kv1.2: Structural Basis of Electromechanical Coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Mildvan, A.; Xia, Z.; Azurmendi, H.; Saraswat, V.; Legler, P.; Massiah, M.; Gabelli, S.; Bianchet, M.; Kang, L.-W.; Amzel, L. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143. [Google Scholar] [CrossRef]
- Perraud, A.-L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a Member of the Nudix Hydrolase Family, Is an Evolutionarily Conserved Mitochondrial ADP-ribose Pyrophosphatase. J. Biol. Chem. 2003, 278, 1794–1801. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.K.; Wu, G.; Massiah, M.A.; Mildvan, A.S. Mutational, Kinetic, and NMR Studies of the Roles of Conserved Glutamate Residues and of Lysine-39 in the Mechanism of the MutT Pyrophosphohydrolase†. Biochemistry 2000, 39, 1655–1674. [Google Scholar] [CrossRef]
- Gabelli, S.B.; Bianchet, A.M.; Ohnishi, Y.; Ichikawa, Y.; Bessman, M.J.; Amzel, L.M. Mechanism of the Escherichia coli ADP-ribose pyrophosphatase, a Nudix hydrolase. Biochemistry 2002, 41, 9279–9285. [Google Scholar] [CrossRef]
- Legler, P.M.; Massiah, M.A.; Mildvan, A.S. Mutational, Kinetic, and NMR Studies of the Mechanism ofE. coliGDP-Mannose Mannosyl Hydrolase, an Unusual Nudix Enzyme†. Biochemistry 2002, 41, 10834–10848. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.W.; Perraud, A.L.; Scharenberg, A.; Stoddard, B.L. The Crystal Structure and Mutational Analysis of Human NUDT9. J. Mol. Biol. 2003, 332, 385–398. [Google Scholar] [CrossRef]
- Tong, L.; Denu, J.M. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubisha, O.; Rafty, L.A.; Takanishi, C.L.; Xu, X.; Tong, L.; Perraud, A.-L.; Scharenberg, A.M.; Denu, J.M. Metabolite of SIR2 Reaction Modulates TRPM2 Ion Channel. J. Biol. Chem. 2006, 281, 14057–14065. [Google Scholar] [CrossRef] [Green Version]
- Nagamine, K.; Kudoh, J.; Minoshima, S.; Kawasaki, K.; Asakawa, S.; Ito, F.; Shimizu, N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 1998, 54, 124–131. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-Ribose and Hydrogen Peroxide Synergize with ADP-Ribose in the Activation of TRPM2 Channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef]
- McHugh, D.; Flemming, R.; Xu, S.-Z.; Perraud, A.-L.; Beech, D.J. Critical Intracellular Ca2+ Dependence of Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel Activation. J. Biol. Chem. 2003, 278, 11002–11006. [Google Scholar] [CrossRef] [Green Version]
- Sano, Y.; Inamura, K.; Miyake, A.; Mochizuki, S.; Yokoi, H.; Matsushime, H.; Furuichi, K. Immunocyte Ca2+ Influx System Mediated by LTRPC2. Science 2001, 293, 1327–1330. [Google Scholar] [CrossRef]
- Starkus, J.; Beck, A.; Fleig, A.; Penner, R. Regulation of TRPM2 by Extra- and Intracellular Calcium. J. Gen. Physiol. 2007, 130, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but in-tracellularly of the gate. J. Gen Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Tóth, B.; Csanády, L. Identification of Direct and Indirect Effectors of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel*. J. Biol. Chem. 2010, 285, 30091–30102. [Google Scholar] [CrossRef] [Green Version]
- Tong, Q.; Zhang, W.; Conrad, K.; Mostoller, K.; Cheung, J.Y.; Peterson, B.Z.; Miller, B.A. Regulation of the Transient Receptor Potential Channel TRPM2 by the Ca2+ Sensor Calmodulin. J. Biol. Chem. 2006, 281, 9076–9085. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Tóth, B.; Szollosi, A.; Chen, J.; Csanády, L. Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. eLife 2018, 7, e36409. [Google Scholar] [CrossRef]
- Iordanov, I.; Tóth, B.; Szollosi, A.; Csanády, L. Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Iordanov, I.; Mihályi, C.; Tóth, B.; Csanády, L. The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. eLife 2016, 5, e17600. [Google Scholar] [CrossRef]
- Tóth, B.; Csanády, L. Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc. Natl. Acad. Sci. USA 2012, 109, 13440–13445. [Google Scholar] [CrossRef] [Green Version]
- Perraud, A.-L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Synthesis and degradation of cyclic ADP-ribose by NAD glycohydrolases. Science 1993, 261, 1330–1333. [Google Scholar] [CrossRef]
- Beck, A.; Kolisek, M.; Bagley, L.A.; Fleig, A.; Penner, R. Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J. 2006, 20, 962–964. [Google Scholar] [CrossRef] [Green Version]
- Lange, I.; Penner, R.; Fleig, A.; Beck, A. Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neu-trophils. Cell Calcium 2008, 44, 604–615. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Liu, Z.; Yu, X.; Ye, P.; Liu, H.; Xue, X.; Yang, L.; Li, Z.; Wu, Y.; Fang, C.; et al. Direct Gating of the TRPM2 Channel by cADPR via Specific Interactions with the ADPR Binding Pocket. Cell Rep. 2019, 27, 3684–3695.e4. [Google Scholar] [CrossRef] [Green Version]
- Heiner, I.; Eisfeld, J.; Warnstedt, M.U.; Radukina, N.; Jüngling, E.; Lückhoff, A. Endogenous ADP-ribose enables calcium-regulated cation currents through TRPM2 channels in neutrophil granulocytes. Biochem. J. 2006, 398, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Tóth, B.; Iordanov, I.; Csanády, L. Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2′-phosphate. J. Gen. Physiol. 2015, 145, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Fliegert, R.; Bauche, A.; Pérez, A.M.W.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2’-Deoxyadenosine 5’-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Moreau, C.; Kirchberger, T.; Swarbrick, J.M.; Bartlett, S.J.; Fliegert, R.; Yorgan, T.; Bauche, A.; Harneit, A.; Guse, A.H.; Potter, B.V.L. Structure-activity relationship of adenosine 5’-diphosphoribose at the transient receptor potential me-lastatin 2 (TRPM2) channel: Rational design of antagonists. J. Med. Chem. 2013, 56, 10079–10102. [Google Scholar] [CrossRef]
- Csanady, L.; Vergani, P.; Gadsby, D.C. Strict coupling between CFTR’s catalytic cycle and gating of its Cl- ion pore revealed by distributions of open channel burst durations. Proc. Natl. Acad. Sci. USA 2010, 107, 1241–1246. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the CFTR Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Clapham, D.E. The TRPM7 Chanzyme Is Cleaved to Release a Chromatin-Modifying Kinase. Cell 2014, 157, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, F.J.P.; Lückhoff, A. Sites of the NUDT9-H Domain Critical for ADP-ribose Activation of the Cation Channel TRPM2. J. Biol. Chem. 2004, 279, 46431–46437. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Dubin, D.T. The role of polyamines in the neutralization of bacteriophage deoxyribonucleic acid. J. Biol. Chem. 1960, 235, 769–775. [Google Scholar] [CrossRef]
- Rafty, L.A.; Schmidt, M.T.; Perraud, A.-L.; Scharenberg, A.M.; Denu, J.M. Analysis of O-Acetyl-ADP-ribose as a Target for Nudix ADP-ribose Hydrolases. J. Biol. Chem. 2002, 277, 47114–47122. [Google Scholar] [CrossRef] [Green Version]
- Tóth, B.; Iordanov, I.; Csanády, L. Putative chanzyme activity of TRPM2 cation channel is Unrelated to pore gating. Proc. Natl. Acad. Sci. USA 2014, 111, 16949–16954. [Google Scholar] [CrossRef] [Green Version]
- Pankiewicz, K.W.; Lesiak, K.; Watanabe, K.A. Efficient Synthesis of Methylenebis(phosphonate) Analogues of P1, P2-Disubstituted Pyrophosphates of Biological Interest. A Novel Plausible Mechanism. J. Am. Chem. Soc. 1997, 119, 3691–3695. [Google Scholar] [CrossRef]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Kühn, F.J.P.; Kühn, C.; Winking, M.; Hoffmann, D.C.; Lückhoff, A. ADP-Ribose Activates the TRPM2 Channel from the Sea Anemone Nematostella vectensis Independently of the NUDT9H Domain. PLoS ONE 2016, 11, e0158060. [Google Scholar] [CrossRef]
- Tóth, B.; Iordanov, I.; Csanády, L. Selective profiling of N- and C-terminal nucleotide-binding sites in a TRPM2 channel. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Roth, B.; Lü, W.; Du, J. Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Huang, Y.; Winkler, P.A.; Sun, W.; Lü, W.; Du, J. Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nat. Cell Biol. 2018, 562, 145–149. [Google Scholar] [CrossRef]
- Henderson, R. From Electron Crystallography to Single Particle CryoEM (Nobel Lecture). Angew. Chem. Int. Ed. 2018, 57, 10804–10825. [Google Scholar] [CrossRef]
- Henderson, R.; Baldwin, J.; Downing, K.; Lepault, J.; Zemlin, F. Structure of purple membrane from halobacterium halobium: Recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. Ultramicroscopy 1986, 19, 147–178. [Google Scholar] [CrossRef]
- Glaeser, R.M. Limitations to significant information in biological electron microscopy as a result of radiation damage. J. Ultrastruct. Res. 1971, 36, 466–482. [Google Scholar] [CrossRef]
- Dubochet, J.; Adrian, M.; Chang, J.-J.; Homo, J.-C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, J.; Van Heel, M. Correspondence analysis of aligned images of biological particles. J. Mol. Biol. 1982, 161, 134–137. [Google Scholar] [CrossRef]
- Frank, J.; Penczek, P.; Grassucci, R.; Srivastava, S. Three-dimensional reconstruction of the 70S Escherichia coli ribosome in ice: The distribution of ribosomal RNA. J. Cell Biol. 1991, 115, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.-C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Armache, J.-P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fu, T.-M.; Zhou, Y.; Xia, S.; Greka, A.; Wu, H. Structures and gating mechanism of human TRPM2. Science 2018, 362, eaav4809. [Google Scholar] [CrossRef] [Green Version]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Morais-Cabral, J.H.; Kaufman, A.; MacKinnon, R. Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 Å resolution. Nat. Cell Biol. 2001, 414, 43–48. [Google Scholar] [CrossRef]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Winkler, P.A.; Huang, Y.; Sun, W.; Du, J.; Lü, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nat. Cell Biol. 2017, 552, 200–204. [Google Scholar] [CrossRef]
- Grimm, C.; Kraft, R.; Sauerbruch, S.; Schultz, G.; Harteneck, C. Molecular and Functional Characterization of the Melastatin-related Cation Channel TRPM3. J. Biol. Chem. 2003, 278, 21493–21501. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Drews, A.; Rizun, O.; Wagner, T.F.J.; Lis, A.; Mannebach, S.; Plant, S.; Portz, M.; Meissner, M.; Philipp, S.E.; et al. Transient receptor potential melastatin 1 (TRPM1) is an ion-conducting plasma membrane channel in-hibited by zinc ions. J. Biol. Chem. 2011, 286, 12221–12233. [Google Scholar] [CrossRef] [Green Version]
- Voets, T.; Nilius, B.; Hoefs, S.; van der Kemp, A.W.; Droogmans, G.; Bindels, R.J.; Hoenderop, J.G. TRPM6 Forms the Mg2+ Influx Channel Involved in Intestinal and Renal Mg2+ Absorption. J. Biol. Chem. 2004, 279, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Xia, R.; Mei, Z.-Z.; Mao, H.-J.; Yang, W.; Dong, L.; Bradley, H.; Beech, D.J.; Jiang, L.-H. Identification of Pore Residues Engaged in Determining Divalent Cationic Permeation in Transient Receptor Potential Melastatin Subtype Channel 2*. J. Biol. Chem. 2008, 283, 27426–27432. [Google Scholar] [CrossRef] [Green Version]
- Huffer, K.E.; Aleksandrova, A.A.; Jara-Oseguera, A.; Forrest, L.R.; Swartz, K.J. Global alignment and assessment of TRP channel transmembrane domain structures to explore func-tional mechanisms. eLife 2020, 9, e58660. [Google Scholar] [CrossRef]
- Mei, Z.-Z.; Mao, H.-J.; Jiang, L.-H. Conserved cysteine residues in the pore region are obligatory for human TRPM2 channel function. Am. J. Physiol. Physiol. 2006, 291, C1022–C1028. [Google Scholar] [CrossRef]
- Yin, Y.; Wu, M.; Hsu, A.L.; Borschel, W.F.; Borgnia, M.J.; Lander, G.C.; Lee, S.-Y. Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rohacs, T.; Nilius, B. Regulation of transient receptor potential (TRP) channels by phosphoinositides. Pflügers Arch. Eur. J. Physiol. 2007, 455, 157–168. [Google Scholar] [CrossRef]
- Yudin, Y.; Lukacs, V.; Cao, C.; Rohacs, T. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 2011, 589, 6007–6027. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, E.; Julius, Y.G.E.C.D.; Cheng, Y.G.Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 2016, 534, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; MacKinnon, R. Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180, 340–347.e9. [Google Scholar] [CrossRef]
- Duan, J.; Li, J.; Zeng, B.; Chen, G.-L.; Peng, X.; Zhang, Y.; Wang, J.; Clapham, D.E.; Li, Z.; Zhang, J. Structure of the mouse TRPC4 ion channel. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Bulkley, D.; Guo, Y.; Zhang, W.; Guo, Z.; Huynh, W.; Wu, S.; Meltzer, S.; Cheng, T.; Jan, L.Y.; et al. Electron cryo-microscopy structure of the mechanotransduction channel NOMPC. Nature 2017, 547, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Le, S.C.; Hsu, A.L.; Borgnia, M.J.; Yang, H.; Lee, S.-Y. Structural basis of cooling agent and lipid sensing by the cold-activated TRPM8 channel. Science 2019, 363, eaav9334. [Google Scholar] [CrossRef]
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nat. Cell Biol. 2018, 559, 580–584. [Google Scholar] [CrossRef]
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nat. Cell Biol. 2018, 559, 575–579. [Google Scholar] [CrossRef]
- Matthies, D.; Dalmas, O.; Borgnia, M.J.; Dominik, P.K.; Merk, A.; Rao, P.; Reddy, B.G.; Islam, S.; Bartesaghi, A.; Perozo, E.; et al. Cryo-EM Structures of the Magnesium Channel CorA Reveal Symmetry Break upon Gating. Cell 2016, 164, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.X.; Armache, J.-P.; Lee, C.; Yang, Y.; Zeng, W.; Mootha, V.K.; Cheng, Y.; Bai, X.-C.; Jiang, Y. Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nat. Cell Biol. 2018, 559, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A., Jr.; Lander, G.C.; Lee, S.-Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubcevic, L.; Herzik, M.A., Jr.; Wu, M.; Borschel, W.F.; Hirschi, M.; Song, A.S.; Lander, G.C.; Lee, S.-Y. Conformational ensemble of the human TRPV3 ion channel. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zubcevic, L.; Hsu, A.L.; Borgnia, M.J.; Lee, S.-Y. Symmetry transitions during gating of the TRPV2 ion channel in lipid membranes. eLife 2019, 8, 8. [Google Scholar] [CrossRef]
- Kühn, F.J.P.; Watt, J.M.; Potter, B.V.L.; Lückhoff, A. Different substrate specificities of the two ADPR binding sites in TRPM2 channels of Nematostella vectensis and the role of IDPR. Sci. Rep. 2019, 9, 4985. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szollosi, A. Two Decades of Evolution of Our Understanding of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel. Life 2021, 11, 397. https://doi.org/10.3390/life11050397
Szollosi A. Two Decades of Evolution of Our Understanding of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel. Life. 2021; 11(5):397. https://doi.org/10.3390/life11050397
Chicago/Turabian StyleSzollosi, Andras. 2021. "Two Decades of Evolution of Our Understanding of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel" Life 11, no. 5: 397. https://doi.org/10.3390/life11050397
APA StyleSzollosi, A. (2021). Two Decades of Evolution of Our Understanding of the Transient Receptor Potential Melastatin 2 (TRPM2) Cation Channel. Life, 11(5), 397. https://doi.org/10.3390/life11050397