
A Structural Approach into Drug Discovery Based on Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
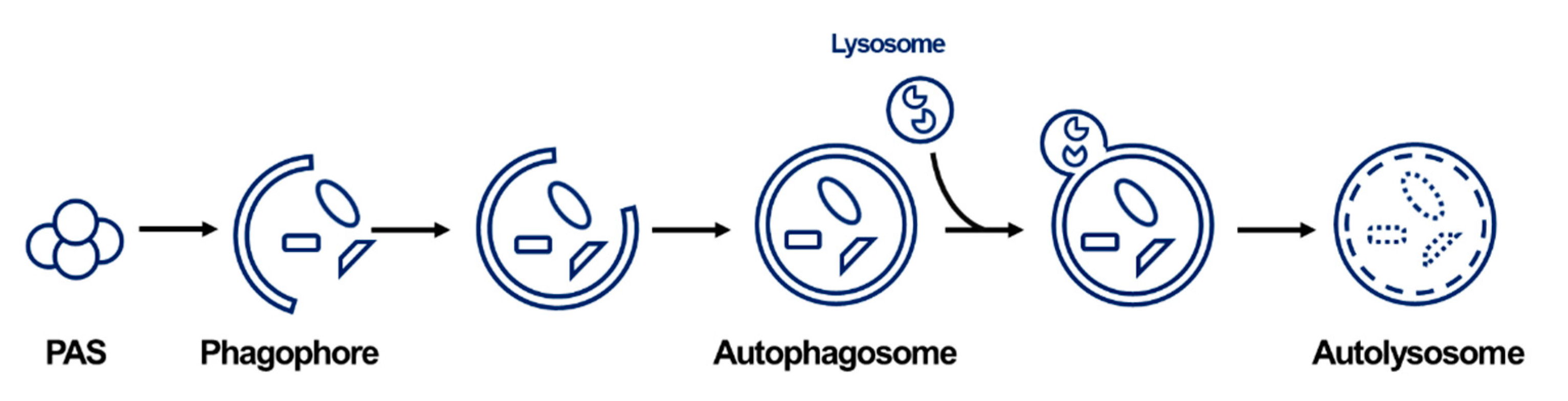
2. General Information on Autophagy
3. Structural Studies of ATG Proteins and Drug Discovery
3.1. ULK1
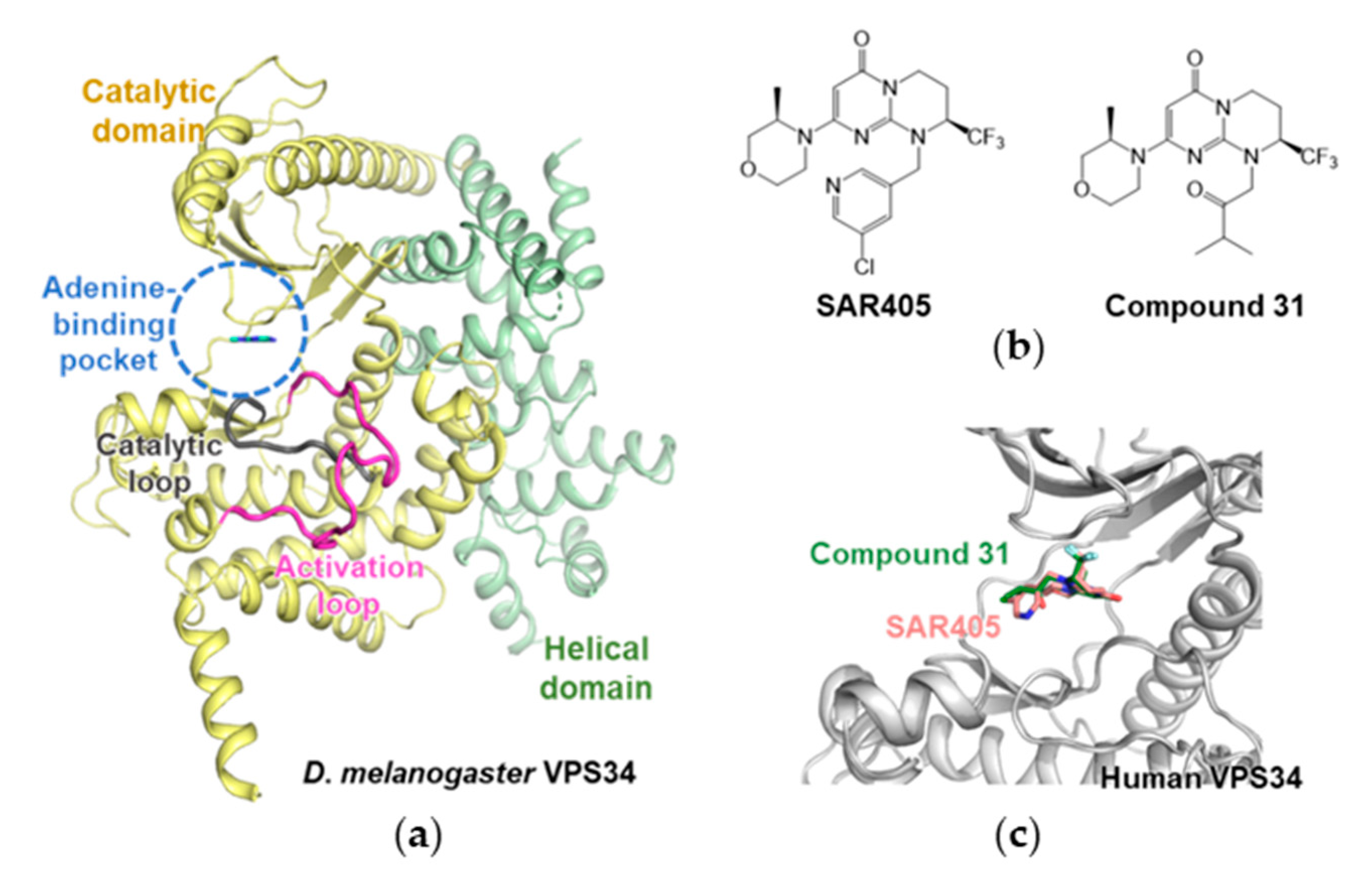
3.2. Autophagy-Specific Lipid Kinase Vacuolar Protein Sorting 34 (VPS34)
3.3. ATG8-ATG3
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klionsky, D.J.; Emr, S.D. Cell biology-autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Zhou, J. Aberrant regulation of autophagy in mammalian diseases. Biol. Lett. 2018, 14, 20170540. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in human diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- He, S.; Li, Q.; Jiang, X.; Lu, X.; Feng, F.; Qu, W.; Chen, Y.; Sun, H. Design of small molecule autophagy modulators: A promising druggable strategy. J. Med. Chem. 2017, 61, 4656–4687. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Chow, K.P.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J. Cell. Mol. Med. 2009, 13, 1371–1380. [Google Scholar] [CrossRef]
- Decaens, T.; Luciani, A.; Itti, E.; Hulin, A.; Roudot-Thoraval, F.; Laurent, A.; Zafrani, E.S.; Mallat, A.; Duvoux, C. Phase II study of sirolimus in treatment-naive patients with advanced hepatocellular carcinoma. Dig. Liver. Dis. 2012, 44, 610–616. [Google Scholar] [CrossRef]
- Yu, H.C.; Lin, C.S.; Tai, W.T.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J. Biol. Chem. 2013, 288, 18249–18259. [Google Scholar] [CrossRef] [Green Version]
- Wilde, L.; Tanson, K.; Curry, J.; Martinez-Outschoorn, U. Autophagy in cancer: A complex relationship. Biochem. J. 2018, 475, 1939–1954. [Google Scholar] [CrossRef]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Perrone, G.; Miura, N.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Santo, L.; Vallet, S.; et al. PI3K/p110δ is a novel therapeutic target in multiple myeloma. Blood 2010, 116, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem. Biol. 2015, 10, 257–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, M.B.; Shokat, K.M. Discovery and structure of a new inhibitor scaffold of the autophagy initiating kinase ULK1. Bioorg. Med. Chem. 2015, 23, 5483–5488. [Google Scholar] [CrossRef] [Green Version]
- Feldman, R.I.; Wu, J.M.; Polokoff, M.A.; Kochanny, M.J.; Dinter, H.; Zhu, D.; Biroc, S.L.; Alicke, B.; Bryant, J.; Yuan, S.; et al. Novel small molecule inhibitors of 3-phosphoinositide-dependent kinase-1. J. Biol. Chem. 2005, 280, 19867–19874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Yang, Z.; Zhen, Y.; Yang, Y.; Chen, Y.; Yuan, Y.; Zhang, L.; Zeng, X.; Chen, L. Discovery of 5-bromo-4-phenoxy-N-phenylpyrimidin-2-amine derivatives as novel ULK1 inhibitors that block autophagy and induce apoptosis in non-small cell lung cancer. Eur. J. Med. Chem. 2020, 208, 112782. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindmo, K.; Stenmark, H. Regulation of membrane traffic by phosphoinositide 3-kinases. J. Cell Sci. 2006, 119, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Takatoh, J.; Wang, F. The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS ONE 2011, 6, e16358. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Bao, Y.; Liu, H.; Kou, X.; Zhang, Z.; Sun, F.; Qian, Z.; Lin, Z.; Li, X.; Liu, X.; et al. VPS34 stimulation of p62 phosphorylation for cancer progression. Oncogene 2017, 36, 6850–6862. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Tavshanjian, B.; Oleksy, A.; Perisic, O.; Houseman, B.T.; Shokat, K.M.; Williams, R.L. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science 2010, 327, 1638–1642. [Google Scholar] [CrossRef]
- Peppard, J.V.; Rugg, C.; Smicker, M.; Dureuil, C.; Ronan, B.; Flamand, O.; Durand, L.; Pasquier, B. Identifying small molecules which inhibit autophagy: A phenotypic screen using image-based high-content cell analysis. Curr. Chem. Genom. Transl. Med. 2014, 8, 3–15. [Google Scholar] [CrossRef]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Pasquier, B.; El-Ahmad, Y.; Filoche-Romme, B.; Dureuil, C.; Fassy, F.; Abecassis, P.Y.; Mathieu, M.; Bertrand, T.; Benard, T.; Barriere, C.; et al. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2H-pyrimido[1,2-a]pyrimidin-6-one: A novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 2015, 58, 376–400. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef]
- Nakatogawa, H.; Ichimura, Y.; Ohsumi, Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007, 130, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duszenko, M.; Ginger, M.L.; Brennand, A.; Gualdron-Lopez, M.; Colombo, M.I.; Coombs, G.H.; Coppens, I.; Jayabalasingham, B.; Langsley, G.; de Castro, S.L.; et al. Autophagy in protists. Autophagy 2011, 7, 127–158. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, K.; Kishi-Itakura, C.; Tsuboi, T.; Sato, S.; Kita, K.; Ohta, N.; Mizushima, N. Autophagy-related Atg8 localizes to the apicoplast of the human malaria parasite Plasmodium falciparum. PLoS ONE 2012, 7, e42977. [Google Scholar] [CrossRef] [Green Version]
- Hain, A.U.; Weltzer, R.R.; Hammond, H.; Jayabalasingham, B.; Dinglasan, R.R.; Graham, D.R.; Colquhoun, D.R.; Coppens, I.; Bosch, J. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. J. Struct. Biol. 2012, 180, 551–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, M.; Noda, N.N.; Nakatogawa, H.; Kumeta, H.; Ohsumi, Y.; Inagaki, F. Autophagy-related protein 8 (Atg8) family interacting motif in Atg3 mediates the Atg3-Atg8 interaction and is crucial for the cytoplasm-to-vacuole targeting pathway. J. Biol. Chem. 2010, 285, 29599–29607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hain, A.U.; Bartee, D.; Sanders, N.G.; Miller, A.S.; Sullivan, D.J.; Levitskaya, J.; Meyers, C.F.; Bosch, J. Identification of an Atg8-Atg3 protein-protein interaction inhibitor from the medicines for Malaria Venture Malaria Box active in blood and liver stage Plasmodium falciparum parasites. J. Med. Chem. 2014, 57, 4521–4531. [Google Scholar] [CrossRef] [Green Version]
- Hain, A.U.; Miller, A.S.; Levitskaya, J.; Bosch, J. Virtual screening and experimental validation identify novel inhibitors of the Plasmodium falciparum Atg8-Atg3 protein-protein interaction. ChemMedChem 2016, 11, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Villa, S.; Legnani, L.; Colombo, D.; Gelain, A.; Lammi, C.; Bongiorno, D.; Ilboudo, D.P.; McGee, K.E.; Bosch, J.; Grazioso, G. Structure-based drug design, synthesis and biological assays of P. falciparum Atg3-Atg8 protein-protein interaction inhibitors. J. Comput. Mol. Des. 2018, 32, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Metlagel, Z.; Otomo, C.; Takaesu, G.; Otomo, T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc. Natl. Acad. Sci. USA 2013, 110, 18844–18849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.S.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.-M.; Kim, D.-H. A Structural Approach into Drug Discovery Based on Autophagy. Life 2021, 11, 526. https://doi.org/10.3390/life11060526
Kang S-M, Kim D-H. A Structural Approach into Drug Discovery Based on Autophagy. Life. 2021; 11(6):526. https://doi.org/10.3390/life11060526
Chicago/Turabian StyleKang, Sung-Min, and Do-Hee Kim. 2021. "A Structural Approach into Drug Discovery Based on Autophagy" Life 11, no. 6: 526. https://doi.org/10.3390/life11060526
APA StyleKang, S. -M., & Kim, D. -H. (2021). A Structural Approach into Drug Discovery Based on Autophagy. Life, 11(6), 526. https://doi.org/10.3390/life11060526